
What is aPAP?
Autoimmune Pulmonary Alveolar Proteinosis, or aPAP, is a disease that belongs to a family of distinct, rare lung diseases collectively referred to as PAP. With aPAP, a buildup of surfactant forms in the alveoli (air sacs) of the lungs. Surfactant is a substance that lines the alveoli and prevents the lungs from collapsing.
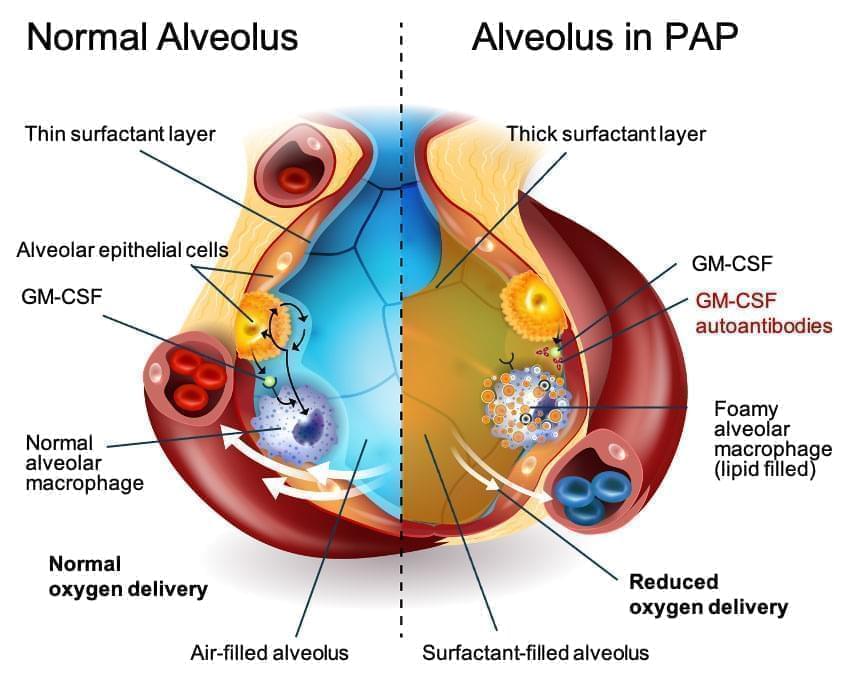
The root cause of aPAP is an autoimmune response against granulocyte-macrophage colony-stimulating factor (GM-CSF). GM-CSF is a naturally occurring protein in the body and is required for immune cells, called macrophages, to properly clear surfactant from the alveoli. In a lung with aPAP, GM-CSF is neutralized by antibodies, rendering macrophages unable to clear surfactant from the alveoli. Surfactant accumulation causes altered gas exchange in the lungs, reduced blood oxygenation, and, ultimately, hypoxemic respiratory failure.
Common symptoms of aPAP
Fatigue
Shortness of breath
Cough
Reduced exercise capacity
Sputum production
The need for an accessible and efficacious medicinal treatment
Today, there are no approved pharmaceutical treatments available for aPAP in the US or Europe. The current standard-of-care is a Whole Lung Lavage (WLL), which does not treat the underlying cause of the disease. WLL is a procedure, performed under general anesthesia, that physically removes surfactant from the lungs. During a WLL, one lung is mechanically ventilated while the other is repeatedly filled with saline. The chest is then percussed (“clapped”) to mix the surfactant with the saline and then drained to remove the saline and surfactant sediment. Due to the percussing, it is not uncommon for patients to experience bruising or cracked ribs following the procedure.
WLL is an invasive and inconvenient procedure that must be performed by highly experienced physicians at specialist sites and necessitates hospitalization and admission to intensive care afterwards. It does not cure aPAP and may only provide temporary symptomatic relief. Because WLL does not treat the underlying cause of aPAP, the lungs refill with surfactant and the procedure may need to be repeated.
There are no formal guidelines for performing WLL in patients with the disease.
Prevalence of aPAP
Autoimmune PAP is a disease that belongs to a family of distinct, rare lung diseases collectively referred to as PAP. Autoimmune PAP represents about 90% of all patients with PAP.
The estimated prevalence of PAP is between 6 and 7 cases per million in the U.S. and Japan, with similar or higher prevalence reported elsewhere in the world.
Japan, a country that has a more centralized approach to diagnosing and treating aPAP, has seen a consistent increase in patients being diagnosed with the disease. It is now estimated the prevalence in Japan could be 26 per million in that country. Read more here.
aPAP occurs in men, women, and children of all ethnic backgrounds and geographic locations, independent of sex, race, and socioeconomic status.
Burden of aPAP
Most patients must learn how to manage the burden of aPAP symptoms and the impacts the disease has on everyday activities.
As a result of surfactant accumulation, gas exchange in the lung is impaired and patients may start to experience shortness of breath and decreased exercise tolerance.
At first, shortness of breath typically occurs upon exertion, but as the disease progresses, it can occur even when a person is at rest. Some patients experience significantly reduced exercise capacity that can dramatically impact the simplest of daily activities, e.g., becoming winded simply walking up a flight of stairs.
Patients may experience cough and episodes of fever—especially if secondary lung infection develops.
In the long-term, the disease can lead to serious complications, including lung fibrosis and the need for lung transplant.
See how Savara is working to change the treatment landscape.
Learn about MOLBREEVI**MOLBREEVI is the FDA and EMA conditionally accepted trade name for molgramostim inhalation solution. It is not approved in any indication.