

Filing
Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, DC 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of
The Securities Exchange Act of 1934
April 27, 2017
Date of Report (Date of earliest event reported)
SAVARA INC.
(Exact name of registrant as specified in its charter)
| Delaware | 001-32157 | 84-1318182 | ||
| (State or other jurisdiction of incorporation) |
(Commission File Number) |
(I.R.S. Employer Identification No.) |
900 Capital of Texas Highway
Las Cimas IV, Suite 150
Austin, Texas 78746
(Address of principal executive offices)
(512) 961-1891
(Registrant’s telephone number, including area code)
Mast Therapeutics, Inc.
3611 Valley Centre Drive, Suite 500
San Diego, California 92130
(Former name or former address, if changed since last report)
Check the appropriate box below if the Form 8-K filing is intended to simultaneously satisfy the filing obligation of the registrant under any of the following provisions (see General Instruction A.2. below):
| ☐ | Written communications pursuant to Rule 425 under the Securities Act (17 CFR 230.425) |
| ☐ | Soliciting material pursuant to Rule 14a-12 under the Exchange Act (17 CFR 240.14a-12) |
| ☐ | Pre-commencement communications pursuant to Rule 14d-2(b) under the Exchange Act (17 CFR 240.14d-2(b)) |
| ☐ | Pre-commencement communications pursuant to Rule 13e-4(c) under the Exchange Act (17 CFR 240.13e-4(c)) |
Indicate by check mark whether the registrant is an emerging growth company as defined in Rule 405 of the Securities Act of 1933 (§ 230.405 of this chapter) or Rule 12b-2 of the Securities Exchange Act of 1934 (§ 240.12b-2 of this chapter).
Emerging growth company ☐
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Table of Contents
Item 2.01. Completion of Acquisition or Disposition of Assets.
On April 27, 2017, Mast Therapeutics, Inc. (the “Company”), completed its business combination with Savara Inc., which changed its name in connection with the transaction to “Aravas Inc.” (“Savara”), in accordance with the terms of the Agreement and Plan of Merger and Reorganization, dated as of January 6, 2017, by and among the Company, Victoria Merger Corp. (“Merger Sub”), and Savara (the “Merger Agreement”), pursuant to which Merger Sub merged with and into Savara, with Savara surviving as a wholly owned subsidiary of the Company (the “Merger”).
Also, on April 27, 2017, in connection with and immediately prior to the effective time of the Merger (the “Effective Time”), the Company (i) effected a reverse stock split at a ratio of one new share for every 70 shares of its common stock outstanding (the “Reverse Stock Split”), and (ii) changed its name to “Savara Inc.” Following the completion of the Merger, the business conducted by the Company became primarily the business conducted by Savara, which is a clinical-stage specialty pharmaceutical company focused on the development and commercialization of novel therapies for the treatment of serious or life-threatening rare respiratory diseases.
Under the terms of the Merger Agreement, the Company issued shares of its common stock to Savara’s stockholders, at an exchange ratio of 0.5860 of a share of common stock (post Reverse Stock Split), in exchange for each share of Savara common stock outstanding as of the Effective Time. The Company also assumed all of the stock options issued and outstanding under the Savara 2008 Stock Option Plan (the “Savara Plan”) and issued and outstanding warrants of Savara, with such stock options and warrants henceforth representing the right to purchase a number of shares of the Company’s common stock equal to 0.5860 multiplied by the number of shares of Savara’s common stock previously represented by such stock options and warrants, as applicable.
Immediately following the Effective Time, there were approximately 15.1 million shares of the Company’s common stock outstanding (post Reverse Stock Split). Immediately following the Effective Time, the former Savara stockholders, warrantholders and optionholders owned approximately 77% of the Company, with the Company’s stockholders, warrantholders and optionholders immediately prior to the Merger, whose warrants, options and shares of the Company’s common stock remain outstanding after the Merger, owning approximately 23% of the Company.
The issuance of the shares of the Company’s common stock to the former stockholders of Savara was registered with the U.S. Securities and Exchange Commission (the “SEC”) on a Registration Statement on Form S-4 (Reg. No. 333-216012) (the “Registration Statement”). The issuance of the shares of the Company’s common stock to holders of stock options issued, or to be issued, under the Savara 2008 Stock Option Plan will be registered with the SEC on a Registration Statement on Form S-8. The Company’s shares of common stock, which were previously listed on NYSE MKT, LLC and traded through the close of business on April 27, 2017 under the ticker symbol “MSTX,” will commence trading on The Nasdaq Capital Market (“Nasdaq”), under the ticker symbol “SVRA” on April 28, 2017. The Company’s common stock has a new CUSIP number, 80511Q 106.
The descriptions of the Merger and Merger Agreement included herein are not complete and are subject to and qualified in their entirety by reference to the Merger Agreement, a copy of which was attached as Exhibit 2.1 to the Company’s Current Report on Form 8-K filed with the SEC on January 9, 2017 and incorporated herein by reference.
On April 27, 2017, the Company issued a press release announcing the completion of the Merger. A copy of the press release is attached hereto as Exhibit 99.1.
Item 2.02 Results of Operations and Financial Condition.
Following the closing of the Company’s business combination with Aravas (formerly known as Savara Inc.) on April 27, 2017 and the payment of severance amounts to former employees and certain transaction related expenses, the Company’s balance of cash, cash equivalents and investment securities was approximately $16 million. This cash balance includes approximately $4 million in aggregate proceeds from the exercise of certain previously issued warrants to purchase Aravas shares and additional capital invested into Aravas prior to the closing of the business combination.
The information in this Item 2.02 shall not be deemed “filed” for purposes of Section 18 of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), or otherwise subject to the liabilities of that Section, nor shall it be incorporated by reference in any filing under the Securities Act of 1933, as amended, or the Exchange Act, except as shall be expressly set forth by specific reference in such filing.
2
Table of Contents
Item 3.03. Material Modification to Rights of Security Holders.
To the extent required by Item 3.03 of Form 8-K, the information contained in Item 2.01 of this Current Report on Form 8-K is incorporated by reference herein.
On April 27, 2017, immediately prior to the Effective Time, the Company amended and restated its certificate of incorporation to (i) effect the Reverse Stock Split and (ii) change the Company’s name to “Savara Inc.” The amendment and restatement of the Company’s certificate of incorporation was approved by the Company’s stockholders at a special meeting of its stockholders on April 27, 2017.
The foregoing descriptions of the Company’s amended and restated certificate of incorporation are not complete and are subject to and qualified in their entirety by reference Company’s amended and restated certificate of incorporation, a copy of which is attached as Exhibit 3.1 hereto and is incorporated herein by reference.
3
Table of Contents
Item 5.01. Changes in Control of Registrant.
The information set forth in Item 2.01 of this Current Report on Form 8-K is incorporated by reference into this Item 5.01.
In accordance with the Merger Agreement, on April 27, 2017, effective as of the Effective Time, Howard C. Dittrich, Peter Greenleaf, and Brian M. Culley resigned from the Board and any respective committees of the Board to which they belonged, Matthew Pauls resigned from his position on the audit committee and nominating and governance committee, and David Ramsay resigned from his position on the nominating and governance committee. The Board appointed, effective as of the Effective Time, Robert Neville, Nevan Elam, Richard J. Hawkins, Joseph S. McCracken and Yuri Pikover as directors of the Company whose terms expire at the Registrant’s next annual meeting of stockholders.
Item 5.02. Departure of Directors or Certain Officers; Election of Directors; Appointment of Certain Officers; Compensatory Arrangements of Certain Officers.
(b) Pursuant to the Merger Agreement, on April 27, 2017, effective as of the Effective Time, Howard C. Dittrich, Peter Greenleaf, and Brian M. Culley resigned from the Board and any respective committees of the Board on which they served, which resignations were not the result of any disagreements with the Company relating to the Company’s operations, policies or practices.
Also, pursuant to the Merger Agreement, on April 27, 2017, effective as of the Effective Time, the Company terminated the employment of Brian M. Culley, the Company’s Chief Executive Officer, Brandi L. Roberts, the Company’s Chief Financial Officer and Senior Vice President, Edwin Parsley, the Company’s Chief Medical Officer and Senior Vice President, and Shana Hood, the Company’s General Counsel, Vice President and Secretary. In connection with the termination of the employment, such officers resigned all of the positions they held with the Company and its subsidiaries.
(c) Effective as of the Effective Time, the Company’s board of directors appointed Robert Neville as the Company’s Chairman and Chief Executive Officer, Taneli Jouhikainen as the Company’s President and Chief Operating Officer, and David Lowrance as the Company’s Chief Financial Officer. There are no family relationships among any of the Company’s directors and executive officers. The information set forth in Item 8.01 of this Current Report on Form 8-K regarding the biographical information, compensation arrangements and related party transaction information for the newly appointed executive officers of the Company is incorporated by reference to this Item 5.02(c). Each of the newly appointed executive officers of the Company entered into the Company’s standard form of indemnification agreement with the Company on April 27, 2017, the form of which is attached hereto as Exhibit 10.12 and incorporated herein by reference.
(d) The information set forth in Item 5.01 of this Current Report on Form 8-K with respect to the appointment of directors to the Company’s board of directors pursuant to and in accordance with the Merger Agreement is incorporated by reference into this Item 5.02(d). The information set forth in Item 8.01 of this Current Report on Form 8-K regarding the related party transaction information for the newly appointed directors of the Company is incorporated by reference to this Item 5.02(d). Each of Nevan Elam, Richard J. Hawkins, Joseph S. McCracken and Yuri Pikover entered into the Company’s standard form of indemnification agreement with the Company on April 27, 2017, the form of which is attached hereto as Exhibit 10.12 and incorporated herein by reference.
4
Table of Contents
Audit Committee
On April 27, 2017, Yuri Pikover and Richard Hawkins were appointed to the audit committee of the Board. David Ramsay will continue to serve as the chairman of the audit committee.
Compensation Committee
On April 27, 2017, Nevan Elam and Joseph McCracken were appointed to the compensation committee of the Board, and Nevan Elam was appointed as the chairman of the compensation committee. Matthew Pauls will continue to serve on the compensation committee.
Nominating and Corporate Governance Committee
On April 27, 2017, Yuri Pikover, Nevan Elam and Joseph McCracken were appointed to the nominating and corporate governance committee of the Board, and Yuri Pikover was appointed as the chairman of the nominating and corporate governance committee.
Item 5.03 Amendments to Articles of Incorporation or Bylaws; Change in Fiscal Year.
(a) To the extent required by Item 5.03 of Form 8-K, the information contained in Item 2.01 and Item 3.03 of this Current Report on Form 8-K is incorporated by reference herein.
Item 5.07 Submission of Matters to a Vote of Security Holders.
On April 27, 2017, the Company held a special meeting of stockholders (the “Special Meeting”) to consider five proposals related to the Company’s previously announced merger with Savara, pursuant to the Merger Agreement. Each of the Company’s proposals was approved by the requisite vote of the Company’s stockholders as described below. The closing of the merger and the related transactions contemplated by the Merger Agreement are currently expected to be completed on or around April 27, 2017.
At the close of business on March 13, 2017, the record date for the Special Meeting, the Company had 254,746,933 shares of common stock issued and outstanding. The holders of a total of 141,518,037 shares of common stock were represented at the Special Meeting by proxy, representing approximately 55.55% of the Company’s issued and outstanding common stock as of the record date, which total constituted a quorum for the Special Meeting in accordance with the Company’s bylaws.
The approval of the Merger Agreement and the issuance of the Company’s common stock pursuant to the Merger Agreement (Proposal No. 1) required the affirmative vote of the holders of a majority of the shares of the Company’s common stock having voting power present in person or represented by proxy at the Special Meeting. The approval of the 1:70 reverse stock split and the change of the Company’s corporate name (Proposal Nos. 2 and 3, respectively) required the affirmative vote of the holders of a majority of the shares of the Company’s common stock having voting power outstanding on the record date for the Special Meeting. The approval of the 1:70 reverse stock split was required in order to authorize the Company’s issuance of the shares of its common stock pursuant to the Merger Agreement and allow for the listing of the common stock of the combined company on the NASDAQ Stock Market following the closing of the merger. As a result, each of Proposal Nos. 1, 2 and 3 were conditioned on each other and, therefore, each was required to pass in order for the merger and the other transactions contemplated by the Merger Agreement to be consummated. The approval, on a non-binding advisory vote basis, of the compensation that will or may become payable by the Company’s to its named executive officers in connection with the merger (Proposal No. 4) and the approval of the adjournment of the Special Meeting, if necessary, to solicit additional proxies (Proposal No. 5) required the affirmative vote of the holders of a majority of the shares of the Company’s common stock having voting power present in person or represented by proxy at the Special Meeting.
The final voting results for each of these proposals is set forth below. Brokers did not have discretionary authority to vote for Proposal Nos. 1, 2, 3 and 4 for the shares of the Company’s common stock held in street name, and as a result, no broker non-votes were received for any of these proposals. For more information on these proposals, please refer to the Company’s prospectus/proxy statement/information statement for the Special Meeting, filed with the Securities and Exchange Commission on March 15, 2017.
5
Table of Contents
Proposal 1. To adopt and approve the Merger Agreement, and to approve the merger and the issuance of the Company’s common stock pursuant to the Merger Agreement:
| 136,743,223 For | 2,208,399 Against | 2,566,415 Abstain | 0 Broker Non-Votes |
Proposal 2. To approve the amended and restated certificate of incorporation of the Company to effect a reverse stock split of the Company’s common stock, at a ratio of one new share for every 70 shares outstanding:
| 131,209,899 For | 6,929,240 Against | 3,378,898 Abstain | 0 Broker Non-Votes |
Proposal 3. To approve the amended and restated certificate of incorporation of the Company to change the name “Mast Therapeutics, Inc.” to “Savara Inc.”:
| 136,330,701 For | 2,328,118 Against | 2,859,218 Abstain | 0 Broker Non-Votes |
Proposal 4. To approve, on a non-binding advisory vote basis, compensation that will or may become payable by the Company to its named executive officers in connection with the merger:
| 112,373,749 For | 23,589,121 Against | 5,555,167 Abstain | 0 Broker Non-Votes |
Proposal 5. To adjourn the Special Meeting, if necessary, to solicit additional proxies if there are not sufficient votes in favor of Proposal Nos. 1, 2, 3 and 4 (although Proposal No. 5 was approved, adjournment of the Special Meeting was not necessary or appropriate because there were sufficient votes at the time of the Special Meeting to approve the other proposals):
| 127,075,022 For | 11,351,993 Against | 3,091,022 Abstain | 0 Broker Non-Votes |
Item 8.01 Other Events.
In connection with the Merger and related transactions described in this Current Report on Form 8-K, the Company provides the following information related to Savara set forth in this Item 8.01.
6
Table of Contents
7
Table of Contents
Cautionary Statement Concerning Forward-Looking Statements
The information in Item 8.01 of this Current Report on Form 8-K, particularly in the sections entitled “Savara Business,” and “Savara Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and the information incorporated herein by reference, include forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. These forward-looking statements are based on current expectations and beliefs and involve numerous risks and uncertainties that could cause actual results to differ materially from expectations. These forward-looking statements should not be relied upon as predictions of future events as we cannot assure you that the events or circumstances reflected in these statements will be achieved or will occur. When used in this report, the words “believe,” “may,” “could,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “expect,” “indicate,” “seek,” “should,” “would” and similar expressions are intended to identify forward-looking statements, though not all forward-looking statements contain these identifying words. All statements, other than statements of historical fact, are statements that could be deemed forward-looking statements.
If any of these risks or uncertainties materializes or any of these assumptions proves incorrect, our results could differ materially from the forward-looking statements in this report. All forward-looking statements in this report are current only as of the date of this report. We do not undertake any obligation to publicly update any forward-looking statement to reflect events or circumstances after the date on which any statement is made or to reflect the occurrence of unanticipated events.
The Company was incorporated in Delaware in December 1995. In October 2000, the Company merged its wholly-owned subsidiary, Biokeys Acquisition Corp., with and into Biokeys, Inc. and changed its name to Biokeys Pharmaceuticals, Inc. In May 2003, the Company merged Biokeys, Inc., a wholly-owned subsidiary, with and into the Company and changed the Company’s name to ADVENTRX Pharmaceuticals, Inc. In March 2013, the Company merged Mast Therapeutics, Inc., a wholly-owned subsidiary, with and into the Company and changed the Company’s name to Mast Therapeutics, Inc. In April 2017, the Company merged its wholly-owned subsidiary, Victoria Merger Corp., with and into Aravas Inc. (formerly, Savara Inc.) and changed the name of the Company to Savara Inc.
8
Table of Contents
Savara is a clinical-stage specialty pharmaceutical company focused on the development and commercialization of novel therapies for the treatment of serious or life-threatening rare respiratory diseases. Savara’s pipeline comprises AeroVanc, a Phase 3 ready inhaled vancomycin, and Molgradex, a Phase 2/3 stage inhaled granulocyte-macrophage colony-stimulating factor, or GM-CSF. Savara’s strategy involves expanding its pipeline of best-in-class products through indication expansion, strategic development partnerships and product acquisitions, with the goal of becoming a leading company in its field. Savara’s management team has significant experience in orphan drug development and pulmonary medicine, in identifying unmet needs, creating and acquiring new product candidates, and effectively advancing them to approvals and commercialization.
AeroVanc, an inhaled formulation of vancomycin, is being developed for the treatment of persistent methicillin-resistant Staphylococcus aureus, or MRSA, lung infection in cystic fibrosis, or CF, patients. CF is a genetic disease that involves sticky mucus buildup in the lungs, persistent lung infections and permanent and progressive respiratory disability. There are approximately 30,000 patients affected by CF in the United States, and MRSA infection has become increasingly common in these patients, with a prevalence of approximately 26%. Persistent MRSA infection in CF patients is associated with increased use of intravenous, or IV, antibiotics, increased hospitalizations, a faster decline of lung function, as well as shortened life-expectancy. Due to the lung pathology associated with CF, persistent MRSA lung infection is difficult to eradicate or manage using oral or IV antibiotics, and there is no standard of care to manage this condition. Whereas inhaled antibiotics have become a cornerstone of treating the most prevalent chronic pathogen in CF patients, Pseudomonas aeruginosa, there are no approved inhaled antibiotics addressing MRSA lung infection. In a randomized, double-blind, placebo-controlled Phase 2 study in CF patients with persistent MRSA infection, AeroVanc met a primary endpoint to reduce MRSA density in sputum, and showed encouraging trends of improvement in lung function, and respiratory symptoms, as well as prolongation of the time to use of other antibiotics, with best responses in subjects under 21 years of age. After receiving detailed guidance from the FDA, Savara has planned a pivotal Phase 3 study of AeroVanc that it anticipates starting in the third quarter of 2017.
Molgradex, an inhaled formulation of recombinant human GM-CSF, is being developed for the treatment of autoimmune pulmonary alveolar proteinosis, or PAP, a rare lung disease characterized by the build-up of lung surfactant in the alveoli, or air sacs, of the lungs. PAP is estimated to have a prevalence of approximately 2,500 patients in the United States. The disease process underlying PAP involves an autoimmune response against a naturally occurring protein, GM-CSF, suppressing the stimulating activity of GM-CSF on lung macrophages which function to clear excess surfactant from the alveoli. The best treatment currently available for PAP is a procedure called whole lung lavage, or WLL, which entails washing out the lungs bronchoscopically with saline, segment by segment, under general anesthesia. By its nature, WLL is an invasive and inconvenient procedure that requires hospitalization, and highly experienced physicians at specialist sites. Based on published investigator-sponsored treatment experience with inhaled GM-CSF, Savara believes Molgradex has the potential to replace the inactivated GM-CSF in PAP patients, and thereby to restore the surfactant clearing activity of the alveolar macrophages, and to become the treatment of choice for PAP. The company has completed a Phase 1 study in healthy volunteers, and is currently conducting a pivotal Phase 2/3 study in Europe and Japan, with top line results expected in the first quarter of 2018.
9
Table of Contents
Savara’s pipeline of product candidates is illustrated in the figure below. In order to fully exploit the potential of its current pipeline, Savara is also pursuing indication expansions of its product candidates, with priority on the development of Molgradex in rare infectious lung diseases, where stimulation of the innate immune system has the potential to improve clinical outcomes. Savara is planning to advance the first such Molgradex indication expansion program into clinical Phase 2 development during 2017, and plans to disclose further information about the program throughout 2017.
Savara’s product candidate pipeline

Savara currently owns exclusive worldwide rights to its product portfolio, except in Japan where rights to Molgradex have been licensed out to Nobelpharma Co., Ltd. AeroVanc has been granted Orphan Drug Designation and Qualified Infectious Disease Product, or QIDP, status for the treatment of persistent MRSA lung infection in CF patients in the United States, and Molgradex has been granted Orphan Drug Designation for the treatment of PAP in the United States and the European Union. The Orphan Drug Designation makes AeroVanc and Molgradex eligible for seven years of exclusivity from approval in the United States, and ten years of exclusivity in the European Union, whereas the QIDP status makes AeroVanc eligible for an additional five years of exclusivity in the United States.
AeroVanc Key Advantages — Savara is currently preparing to initiate a Phase 3 clinical study of AeroVanc, to be conducted primarily in the United States and Canada. Savara has received detailed guidance from the FDA on the design of the study, and believes that the planned study is in accordance with the FDA’s requirements for a sole pivotal study to be submitted for NDA approval. Savara anticipates initiating the study in the third quarter of 2017. Savara believes the results from its Phase 2 study, illustrated in part below, support the use of the same key endpoints and advancing the development of AeroVanc into a larger pivotal Phase 3 study. Notably, the Phase 2 study demonstrated a trend of clinically meaningful improvement in FEV1, a common measure of lung function illustrated below on the left, as well as in time to use of another antibiotic for respiratory infection, illustrated below on the right. The planned primary efficacy endpoint of the Phase 3 study is change from baseline in FEV1, and the primary analysis population will comprise patients under the age of 21, in line with experience from earlier clinical studies of inhaled anti-pseudomonal antibiotics in CF.
10
Table of Contents
Change from baseline in FEV1 (left) and Time to use of other antibiotic for respiratory infection (right)
|
|

| |
| Per Protocol Population, 32 mg dose cohort, < 21 years of age, n = 16 | Intent-to-treat Population, 32 mg dose cohort, < 21 years of age, n = 20 | |
Savara believes that AeroVanc has a number of important characteristics that contribute to its clinical profile and clinical data to date, and that facilitate its regulatory approval and successful commercialization. Specifically, AeroVanc offers:
| • | Strong product foundation, applying a previously approved active substance and previously approved drug delivery technologies. |
| • | High concentration of antibiotic is delivered directly to the lungs, the primary site of infection, which Savara believes can result in higher clinical efficacy and reduced systemic toxicity, as compared with oral or IV delivery of antibiotics. |
| • | Capsule based powder inhaler providing a fast and convenient method of administration, which is very desirable in the CF population, who have a high treatment burden. |
| • | Eligible for strong market protection via orphan drug status, QIDP status, a formulation patent, and an exclusive device supply agreement. |
Molgradex Key Advantages — Savara is currently conducting a Phase 2/3 clinical study, which is referred to as the IMPALA study, of Molgradex in Europe and Japan. Savara has received guidance from the European Medicines Agency, or EMA, on the design of the study, and believes the ongoing study is in accordance with the EMA’s requirements for a sole pivotal study to be used in a marketing authorization application submission in the European Union. Savara anticipates reporting top-line results from the study in the first quarter of 2018. Savara is also in discussions with the FDA to receive guidance on the clinical study requirements for an NDA submission in the United States. Savara expects to have clarity on those requirements later this year. The options include expanding and modifying the ongoing IMPALA study as the sole pivotal study, or conducting a second pivotal study for US regulatory purposes.
Building upon the published investigator-sponsored treatment experience with inhaled GM-CSF, Savara believes Molgradex has the potential to become the treatment of choice for PAP. Molgradex has the following characteristics that Savara believes will contribute to its clinical profile, as well as facilitate its regulatory approval and successful commercialization. Specifically, Molgradex offers:
| • | Strong product foundation, applying a previously approved active substance class and previously approved drug delivery technology. |
| • | GM-CSF is delivered directly to the lungs, the primary site of macrophage function deficiency, which Savara believes can result in high clinical efficacy with limited systemic adverse effects. |
| • | High efficiency nebulizer providing a fast and convenient method of administration, which is highly desirable for long-term treatment in a chronic disease, such as PAP. |
11
Table of Contents
| • | Eligible for strong market protection via orphan drug status, a proprietary cell bank used in the production of the drug substance, and an exclusive device supply agreement. |
Savara’s goal is to become a leading specialty pharmaceutical company focused on treatments for rare respiratory diseases, through the development and commercialization of novel and best-in-class therapeutics to address unmet medical needs in its field. The key elements of Savara’s strategy include:
| • | Pursue AeroVanc and Molgradex indication expansion. While Savara’s immediate priority is to obtain regulatory approvals in the primary indications described above, Savara believes both AeroVanc and Molgradex have the potential to be used for the treatment of several other diseases. In particular, Savara is exploring the use of Molgradex for the treatment of certain rare infectious lung diseases. |
| • | Expand the product pipeline through strategic product acquisitions. In addition to broadening its current pipeline through indication expansion, Savara’s strategy includes expansion of its product pipeline through strategic partnerships and product acquisitions, such as its acquisition of the Molgradex program through the asset purchase of Serendex Pharmaceuticals in 2016. A key priority has been to exploit known chemical entities or classes in novel ways, such as delivery of drug directly into the lungs, for the treatment of serious or life-threatening lung diseases. While Savara has developed an internal core competence in inhaled drug development, the company is technology agnostic. Future pipeline expansion decisions will be based on the unmet medical need within a specific disease, the commercial opportunity, and the ability to rapidly develop and commercialize a product candidate. |
| • | Operate by outsourcing capital intensive operations. Savara plans to continue to pursue the development and manufacturing of its product candidates by outsourcing most clinical development and all manufacturing operations. Savara’s business model has facilitated rapid development of its pipeline by using high quality specialist vendors and consultants in a capital efficient manner. |
| • | Establish its own sales and marketing capabilities to commercialize its products in the United States. Savara plans to commercialize its pipeline through its own specialty salesforce or strategic marketing partnerships in the United States. Outside the United States, Savara plans to commercialize its products in collaboration with partners that have the resources and infrastructure to successfully commercialize Savara’s innovative therapeutics. |
Overview of AeroVanc
Background on MRSA infection in cystic fibrosis
CF is a genetic disease characterized, in part, by the prevalence of thick, sticky mucus produced in the lung, frequent lung infections, and a resultant decline in pulmonary function. As the disease progresses, patients’ lungs are typically infected with bacteria that are difficult to eradicate. Inhaled antibiotics, including tobramycin (TOBI, Novartis AG), and aztreonam (Cayston, Gilead Sciences), have become a cornerstone of the treatment of the most common chronic pathogen, Pseudomonas aeruginosa, in order to control the infection and improve lung function and quality of life. In recent years, MRSA lung infection has become increasingly common in CF, with a prevalence of 26 % according to the most recent (2015) data report of the Cystic Fibrosis Foundation. Importantly, persistent MRSA lung infection has been associated with worse clinical outcomes in CF, including a faster decline of lung function1 and a shorter life expectancy.2 The increasing prevalence and high clinical impact of MRSA infection in CF have created an unmet need for improved therapies to help address the condition. Considering the established practice of treating chronic Pseudomonas aeruginosa infection in CF using inhaled
| 1 | Dasenbrook EC, Merlo CA, Diener-West M, et al. “Persistent Methicillin-resistant Staphylococcus aureus and Rate of FEV1 Decline in Cystic Fibrosis.” Am J Respir Crit Care Med 2008;178, 814-821. |
| 2 | Dasenbrook EC, Checkley W, Merlo CA, et al. “Association Between Respiratory Tract Methicillin-Resistant Staphylococcus aureus and Survival in Cystic Fibrosis.” JAMA 2010;303, 2386-2392 |
12
Table of Contents
antibiotics, all of which have limited activity against MRSA, it would be logical to attempt treatment of chronic MRSA infection with an inhaled antibiotic active against MRSA. Savara believes that AeroVanc is the first inhaled antibiotic being developed to specifically treat MRSA infection of the lungs.
Current MRSA treatment options in CF
Persistent MRSA lung infection in CF patients is difficult to eradicate or manage using oral or IV antibiotics, and there is currently no standard of care to manage the infection in CF patients despite the high need.3 In contrast to the established treatment of Pseudomonas aeruginosa infection with inhaled antibiotics, there is no FDA-approved inhaled antibiotic treatment available for MRSA infection.
IV vancomycin or linezolid are the most commonly used drugs for the treatment of acute pulmonary exacerbation in CF patients with MRSA infection, and they may be used in combination with other IV antibiotics in patients with simultaneous Gram-negative infections, such as Pseudomonas aeruginosa. For MRSA lung infection, vancomycin is available only in IV form, and while highly effective against MRSA and other Gram-positive bacteria, chronic home-based use of IV vancomycin is not practical, and chronic use has also been associated with systemic toxicity, especially renal toxicity and ototoxicity.
According to research conducted by Savara, there is increasing clinical need to treat chronic MRSA infection in CF. In the absence of an inhaled antibiotic, there is emerging use of oral anti-MRSA antibiotics in an attempt to suppress the MRSA infection, and in hope of reducing the occurrence of acute pulmonary exacerbations. In a survey conducted by Savara, 27 % of the surveyed CF specialists in the US regularly utilize antibiotics targeting MRSA as a suppressive treatment (any dosage form) in patients with frequent exacerbations or other symptoms for which MRSA is considered a cause or contributing factor. This practice is emerging despite the absence of established consensus or guidelines relating to the use of oral anti-MRSA antibiotics in CF, or evidence of efficacy established in controlled studies.
As with current inhaled anti-pseudomonal drugs, Savara believes that there is significant clinical advantage in delivering an anti-MRSA antibiotic, such as vancomycin, directly to the site of infection to maximize the clinical efficacy, reduce systemic exposure and the risk of adverse effects, and to enable convenient use of the product outside of the hospital setting. The aerosolized IV form of vancomycin, administered by nebulization, has been used in multiple small published clinical studies, mainly to treat ventilator-associated pneumonia in an intensive care setting. In these studies and case reports, nebulized vancomycin had good antibacterial efficacy and was generally well tolerated. In recent years, according to interviews conducted by Savara, many of the leading CF centers in the United States have explored the use of inhaled vancomycin to treat MRSA infected CF patients on a chronic basis, by nebulizing the IV form of vancomycin. The experience gained from this type of treatment has been encouraging, and provides anecdotal reports of the safety and clinical utility of inhaled vancomycin for periods exceeding many years in some patients. Similarly, in the 1990’s, nebulized IV tobramycin was explored as a treatment of Pseudomonas aeruginosa infections in CF patients. This experience stimulated the development of TOBI®, which has become the most widely used inhaled antibiotic worldwide, and a cornerstone of chronic treatment of Pseudomonas aeruginosa lung infection in CF.
Savara believes that inhaled antibiotics, as well as other palliative treatments, will continue to have a central role in the management of CF. Various disease modifying drugs, such as CF Transmembrane Conductance Regulator (CFTR) modulators, that attempt to address the underlying cause of CF, i.e. to restore or improve the function of the CFTR protein that is defective or dysfunctional in CF patients, have recently been launched. Whereas these disease-modifying drugs on average result in modest improvement in lung function and potentially slower rate of lung function decline, patients on these drugs continue to have chronic infections that require antibiotic treatment, and their lung function continues to decline.
| 3 | Zobell JT, Epps KL, Young DC, Montague M, Olson J, Ampofo K, Chin MJ, Marshall BC, Dasenbrook E. “Utilization of antibiotics for methicillin-resistant Staphylococcus aureus infection in cystic fibrosis.” Pediatric Pulmonology (June 2015) Volume 50, Issue 6, pages 552–559 |
13
Table of Contents
AeroVanc Product Description
AeroVanc, or Vancomycin Hydrochloride Inhalation Powder, is a novel inhaled formulation of vancomycin being developed for the treatment of persistent MRSA lung infection in patients with CF. Vancomycin is a glycopeptide antibiotic that was discovered in the mid-1950’s and is commonly used in the prophylaxis and treatment of infections caused by Gram-positive bacteria. Vancomycin acts by inhibiting proper cell wall synthesis of aerobic and anaerobic Gram-positive bacteria, and is generally not active against Gram-negative bacteria.
AeroVanc consists of a capsule dosage form containing a proprietary dry powder formulation of vancomycin hydrochloride intended for oral inhalation with the AeroVanc inhaler. The AeroVanc inhaler is a commercialized, hand-held, manually operated, breath-activated device.
Savara anticipates that AeroVanc will be used predominantly to suppress chronic MRSA lung infection, which has the potential to improve patients’ lung function and respiratory symptoms, and to prolong the time to pulmonary exacerbation and need of systemic antibiotics. AeroVanc is not intended to replace IV vancomycin or other IV antibiotics in the treatment of acute pulmonary exacerbations associated with MRSA. However, chronic AeroVanc use has the potential to reduce the occurrence of these exacerbations, and thereby the need for IV treatments and hospitalizations.
Savara believes there will be broad adoption of AeroVanc in CF once available based on a high level of interest for the product from direct clinician surveys, as well as market research of key opinion leaders in the field of CF. Notably, a clear majority (94 %) of the surveyed CF physicians in the United States would expect to prescribe AeroVanc to their patients with MRSA lung infection, if approved by the FDA. Likewise, according to payer interviews conducted in the United States, an AeroVanc launch would receive reimbursement support given the high unmet need in an orphan indication and a current lack of comparable products.
Clinical Development of AeroVanc
Phase 3
Savara intends to initiate a Phase 3 clinical study designed to demonstrate the safety and efficacy of AeroVanc in CF patients with persistent MRSA lung infection. The plan is to initiate this trial in the third quarter of 2017. The study is planned to be conducted primarily in the United States and Canada.
Savara has received detailed guidance from the FDA on the design of the study, and believes that the planned study is in accordance with the FDA’s requirements for a sole pivotal study to be used in an NDA submission. The study has also been planned in consultation with the Cystic Fibrosis Foundation’s Therapeutic Development Network. The Phase 3 study is designed to detect whether the administration of AeroVanc results in a significant improvement in lung function. The study will assess a 32 mg dose administered twice a day for three on/off cycles of 28 days. The planned primary efficacy endpoint is absolute change from baseline in FEV1 percent predicted, a commonly used measure of lung function. Other efficacy endpoints include the time to use of other antibiotics for pulmonary infection, and a respiratory symptom score.
The planned Phase 3 study is a randomized (1:1), double-blind, placebo-controlled study of AeroVanc in approximately 200 CF patients with persistent MRSA lung infection. The plan is to enrich the study with younger patients, by enrolling 75 % of the subjects between the ages of 6 and 21 years. This was the population most responsive to treatment in the Phase 2 study, and will form the primary analysis population of the study. The duration of the study drug (AeroVanc or placebo) administration will be three cycles of 28 days on drug and 28 days off drug, during which time the primary efficacy endpoint will be measured and assessed. Following the efficacy study period, subjects will transition into another three cycles (28 days on treatment, 28 days off treatment per cycle) of open label AeroVanc use to provide more information on long-term safety.
14
Table of Contents
The planned primary efficacy endpoint of the study is the mean absolute change from baseline in FEV1 percent predicted. In accordance with guidance from the FDA, the endpoint will be analyzed sequentially at Week 4 (first treatment cycle), and at Week 20 (third treatment cycle). Both time points will be tested at a statistical significance level of p = 0.05 due to the sequential nature of the analysis. Savara believes that a statistically significant improvement at Week 20 would provide support for a chronic treatment label, whereas improvement at Week 4 only may result in a more restricted label. Approval in any form is subject to the positive evaluation of the clinical meaningfulness of the treatment effect, judged by the review of all data, including safety data, and the outcome of key secondary endpoints, such as time to use of other antibiotics.
In the single-cycle Phase 2 study, with missing data imputed using conservative rules adopted by the FDA, a difference in the mean absolute change in FEV1 percent predicted of 4.3 % was observed between the treatment arms in subjects below 21 years of age. Based on the observed treatment effect size and variability, a sample size of 45 subjects per arm would provide 90 % power to detect a statistically significant difference at an alpha level of 0.05. To account for a potential loss of power caused by premature discontinuations in a three-cycle study, a sample size of 75 subjects per arm will be enrolled.
Selection of the dose for the study was made based on the Phase 2 study in CF patients. In that study, administration of the 32 mg bid dose resulted in sputum trough vancomycin concentrations that were on average more than 100-fold above the observed minimum inhibitory concentration (MIC90) value, suggesting that the concentrations reached after repeated administration of the 32 mg bid dose are likely to be sufficient for effective management of MRSA infection. In terms of safety and tolerability, the 32 mg AeroVanc dose did not appear significantly different from placebo, and produced encouraging trends of efficacy in all key endpoints in subjects below 21 years of age. In contrast, the higher AeroVanc dose of 64 mg bid was not as well tolerated in the older subjects (above 21 years of age), resulting in an increased number of premature discontinuations of the study drug treatment in this subgroup.
After the completion of the Phase 3 study, Savara intends to submit an NDA applying the 505(b)(2) regulatory pathway. In addition to being designated an Orphan Drug Product and QIDP, AeroVanc has been designated a Fast Track development program by the FDA.
Completed Clinical Studies
Phase 1
In a Phase 1 single escalating dose study, AeroVanc was shown to be generally well tolerated and safe, with a favorable pharmacokinetic profile. In the study, AeroVanc inhalation powder was administered to 18 healthy volunteers (doses of 16 mg, 32 mg, and 80 mg), and seven patients with CF (doses of 32 mg, and 80 mg). AeroVanc demonstrated a relatively slow pulmonary absorption phase (tmax of 1.33 h — 2.08 h), followed by distribution and elimination comparable to IV administration. The mean absolute bioavailability across all AeroVanc doses was 49 % (SD 8 %), with no apparent differences observed between the doses. The absolute bioavailability closely corresponds with the pulmonary absorption of vancomycin, considering that vancomycin is not absorbed from the gastrointestinal tract. The mean Cmax of AeroVanc after an 80 mg dose was 618 ng/mL, corresponding to approximately one fifth of the dose adjusted Cmax after a 250 mg dose of IV vancomycin. The dose linearity of AeroVanc in terms of Cmax and AUC values was excellent (R2 > 0.99). In the CF patients, all subjects had sputum vancomycin concentrations in high excess of the minimum inhibitory concentration, or MIC, of vancomycin for MRSA (2 µg/mL) at one hour after the administration of AeroVanc with both the 32 mg and the 80 mg dose (mean of 106 µg/mL, and 261 µg/mL, respectively). At later time points, the concentrations decreased, but on average remained above the MIC values for up to 24 hours. Variability in sputum concentrations was high, as expected.
All adverse events in the healthy volunteers were classified as mild, and all events that were considered probably drug-related involved local irritation effects and resolved spontaneously and rapidly (between 15 and 60 minutes).
15
Table of Contents
Small reduction in the post-dose FEV1 (7 % — 11 %) was observed in three subjects after the 80 mg dose. None of the subjects required bronchodilator treatment, and the changes were considered by the independent Drug Safety Monitoring Board to be clinically non-significant. In CF patients, chest congestion and/or chest tightness were reported by four of the seven patients, and there appeared to be a slight trend towards more adverse events at the higher dose (80 mg). All reported respiratory adverse events were mild, none of the patients felt distressed, and the events either did not require treatment or resolved after airway clearance and/or albuterol inhalation. Based on the sputum concentration data, dose levels of 32 mg and 64 mg twice a day were selected for use in the Phase 2 study.
Phase 2
In a Phase 2 clinical study in CF patients with persistent MRSA lung infection, AeroVanc met a primary endpoint to reduce MRSA density in sputum, and showed encouraging trends of improvement in lung function, prolongation of the time to use of other antibiotics, and respiratory symptoms, with best responses in subjects below 21 years of age. Savara believes that the consistency of the responses across the different endpoints, as well as the magnitude of change in the younger subjects, supports advancing the product into a Phase 3 clinical study. The results of the Phase 2 study have been summarized and presented to the FDA in an End of Phase 2 Meeting, and the FDA has subsequently given Savara detailed guidance on the design and analysis of a Phase 3 study, as presented above in section “Phase 3”. The key findings of the Phase 2 study are described below.
The study was a randomized, double-blind, placebo-controlled study in 87 CF patients with persistently positive MRSA culture from their sputum samples. The Phase 2 study consisted of a 28-day AeroVanc treatment at a dose level of 32 mg bid or 64 mg bid, with an eight-week follow-up. The study was conducted at 40 sites in the United States. Quantitative MRSA cultures from spontaneously expectorated sputum samples were used as the primary endpoint of the study. The average baseline values in both active drug cohorts, as well as the placebo cohorts were high, ranging from 6.78 to 7.65 log10 CFU/mL. In the primary endpoint analyses (MITT population), a reduction from baseline in MRSA CFU was observed in the 32 mg and 64 mg dose cohorts pooled compared to placebo by -0.52 log10 CFU/mL and -0.06 log10 CFU/mL for the AeroVanc and placebo doses, respectively (p = 0.0312); in the 64 mg dose cohort by -0.63 log10 CFU/mL and 0.16 log10 CFU/mL for the AeroVanc and placebo doses, respectively (p = 0.0145); in the 32 mg dose cohort by -0.25 log10 CFU/mL and -0.30 log10 CFU/mL for the AeroVanc and placebo doses, respectively (p = 0.8352).
MICs of vancomycin for MRSA cultured from the sputum samples were determined using a broth microdilution technique at baseline, at each visit during the administration of AeroVanc, as well as at the post-administration follow-up time points. The distribution of MIC values was very narrow, with the MIC50 and MIC90 both at 0.5 µg/mL at baseline. At baseline, all strains were susceptible to vancomycin, with MIC values £ 1 µg/mL, and there were no notable changes in the MIC distribution at any of the time points following the baseline sample, suggesting the susceptibility of MRSA to vancomycin was not affected by the 28 days of pulmonary administration of AeroVanc.
As illustrated in the graph below, vancomycin peak and trough concentrations in sputum at Day 8 and Day 29 were in high excess over the generally accepted level of MIC (mean Ctrough/MIC ratio > 35) after multiple dosing in all subjects at both dose levels, with apparent dose-dependency, but no notable difference in Ctrough between the two time points. The generally accepted MIC of vancomycin for MRSA is illustrated below by the dotted line, at 2 µg/mL.
16
Table of Contents
Vancomycin sputum concentrations after administration of AeroVanc at various time points

In terms of safety, the most frequent adverse events reported were related to the respiratory system. The AeroVanc 32 mg bid dose was well tolerated, with no significant difference in adverse events as compared with placebo. However, a higher incidence of adverse events, most frequently consistent with signs and symptoms of bronchoconstriction, and a significantly higher rate of premature study drug discontinuations were seen in adult patients with the 64 mg bid AeroVanc dose, as compared with placebo and the 32 mg AeroVanc dose. The discontinuations were most commonly reported to be due to drug intolerability (mainly bronchoconstriction and/or chest tightness) or pulmonary exacerbation, and typically occurred within the first two weeks from the start of drug administration.
Based on the observed clinical results in the 32 mg cohort of subjects below 21 years of age, the observed high vancomycin concentrations in sputum at both dose levels, and the high discontinuation frequency in adult subjects at the 64 mg dose, the Phase 3 study is planned to be conducted using the 32 mg dose, and will focus enrollment on subjects below 21 years of age. Accordingly, the key Phase 2 data from this cohort, below 21 years of age, are summarized below.
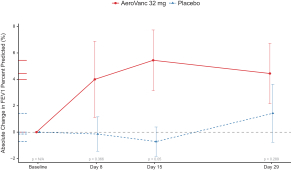
To assess effects of AeroVanc on lung function, absolute change in FEV1 percent predicted from baseline was measured at each study visit. While AeroVanc reduced MRSA density in sputum, the change in FEV1 compared with placebo did not reach statistical significance in subjects of all ages. Notably, post hoc analyses identified encouraging improvement in FEV1 in subjects 21 years of age or younger, consistently across all time points during the treatment period, as illustrated below. The mean absolute change in FEV1 percent predicted observed in the AeroVanc arm is considered clinically meaningful, with an improvement ranging between 4 % and 6 % (or 6 % and 10 % on a relative change basis). In this subgroup, the difference between AeroVanc and placebo was statistically significant at the 2-week time point (p = 0.05). A mean reduction of 0.8 log10 CFU/mL from baseline in MRSA CFUs, the primary endpoint, was also observed after 28 days of AeroVanc administration in subjects below 21 years of age, as illustrated below, the difference between AeroVanc and placebo being statistically significant (p = 0.05).
17
Table of Contents
Change from baseline in FEV1
(Per Protocol Population, 32 mg dose cohort, below 21 years of age, n = 16)

These results are consistent with previous studies using inhaled tobramycin (TOBI® or TOBI Podhaler®) for the treatment of P. aeruginosa infection in CF, where improvement in FEV1 was predominantly seen in younger subjects4. In the early TOBI trials, reported in the 1990’s, during an era when the use of inhaled antibiotics was not yet prevalent, children and adolescents (below 18 years of age) showed relative improvements of greater than 14 % as compared with only 6 % in adults5. However, in more recent studies, reported in 2012, the relative FEV1 improvements have been considerably smaller, either being absent or less than 2 % in adults.6
As illustrated below, a mean reduction of 0.8 log10 CFU/mL from baseline in MRSA CFUs, the primary endpoint, was observed after 28 days of AeroVanc administration in subjects below 21 years of age, the difference between AeroVanc and placebo being statistically significant (p = 0.05).
| 4 | Weers J. “Inhaled antimicrobial therapy – Barriers to effective treatment. Advanced Drug Delivery Reviews.” (2015): 24-43. |
| 5 | Ramsey BW, Pepe MS, Quan JM, Otto KL, Montgomery AB, Williams-Warren J, Vasiljev-K M, Borowitz D, Bowman CM, Marshall BC, Marshall S, Smith AL. “Intermittent administration of inhaled tobramycin in patients with cystic fibrosis. Cystic Fibrosis Inhaled Tobramycin Study Group.” New England Journal of Medicine. 1999 Jan 7;340(1):23-30. |
| 6 | TOBI Podhaler SBA; NDA-201688, 2012 |
18
Table of Contents
Change in MRSA density in sputum
(Intent-to-treat Population, 32 mg dose cohort, below 21 years of age, n = 20)

A greater reduction in CFRSD-CRISS, the respiratory symptom score, was observed in the below 21-year age group consistently at all time points, as compared with placebo, but the difference was not statistically significant.
A trend of prolongation of the time to use of another antibiotic for respiratory symptoms was observed in the AeroVanc arm of the 32 mg dose cohort, as compared with placebo, illustrated below. Whereas in this single cycle study several subjects in the AeroVanc arm were prescribed other antibiotics at the scheduled one-month post-treatment visit (approximately Day 56), such treatment would not be expected to be prescribed during chronic AeroVanc treatment, or in a multiple-cycle study, because the timing would coincide with the start of a new AeroVanc treatment period.
19
Table of Contents
Time to use of other antibiotics for respiratory infection
(Intent-to-treat Population, 32 mg dose cohort, below 21 years of age, n = 20)

In summary, AeroVanc reduced MRSA density in sputum, and showed encouraging trends of improvement in lung function, prolongation of the time to use of other antibiotics, and respiratory symptom, with best responses in subjects below 21 years of age. Savara believes that the consistency of the responses across the different endpoints, as well as the magnitude of change in the younger subjects, supports advancing the product into a Phase 3 clinical study.
Human factor study
Savara has performed a human factor study to better understand patient reactions to the AeroVanc inhaler device, the drug capsule and written instructions. 14 CF patients, representing a variety of sex, ethnicity and dominant hand preference and ranging in age from 12 to 56 years participated in the study. Patients were given the device, capsules and instructions to simulate use (no drug) and provide feedback. In summary, all patients were able to use the device properly and no device design issues were identified that could impact proper use.
Overview of Molgradex
Background on PAP
PAP is a rare lung disease, which affects up to seven out of a million people in the United States7, and has a similar prevalence in Japan8. PAP is characterized by the build-up of lung surfactant in the alveoli, or air sacs, of
| 7 | Trapnell BC, Avetisyan R, Carey B, Zhang W, Kaplan P, Wang H. Prevalence of pulmonary alveolar proteinosis (PAP) determined using a large health care claims database. Am J Respir Crit Care Med. 2014;VOL:abstract A6582. |
| 8 | Inoue Y, Trapnell BC, Tazawa R, Arai T, Takada T, Hizawa N et al. Characteristics of a large cohort of patients with autoimmune pulmonary alveolar proteinosis in Japan. Am J Respir Crit Care Med 177: 752–62, 2008 |
20
Table of Contents
the lungs. The surfactant consists of proteins and lipids, and is an important physiological substance that coats the inside of the alveoli to prevent the lungs from collapsing. The lungs continuously produce new active surfactant. In a healthy lung, the old and inactivated surfactant is cleared and digested by immune cells called alveolar macrophages. In PAP lungs, however, the macrophages fail to clear the surfactant from the alveoli, leading to gradual accumulation of excess surfactant in the alveoli. The root cause of PAP is an autoimmune response against a naturally occurring protein of the body, GM-CSF. Pulmonary macrophages need to be stimulated by GM-CSF to function properly, but in autoimmune PAP, GM-CSF is deactivated by antibodies against GM-CSF, rendering the macrophages unable to perform their tasks, such as clearing the surfactant from the alveoli.
PAP commonly affects men in early middle age, but both sexes and subjects of any age can be affected. As a result of the accumulation of excess surfactant, gas exchange in the lungs is obstructed, and patients start to experience shortness of breath, and decreased exercise tolerance. Shortness of breath is typically first observed upon exertion, but as the disease progresses, also at rest. Patients may experience chronic cough, as well as episodes of fever, chest pain, or coughing blood, especially if secondary lung infection develops. In the long term, the disease can lead to serious complications, including lung fibrosis and the need for lung transplant. Mortality due to PAP has decreased over the last decades with better clinical management, but in rare cases serious lung infections or respiratory insufficiency may lead to death.
Current treatment options of PAP
The current standard of care for PAP is a procedure called whole lung lavage, or WLL, which entails washing out the lungs with saline under general anesthesia. WLL is an invasive and inconvenient procedure that requires highly experienced physicians at specialist sites. The procedure in conducted in an operating room, thereby requiring hospitalization, and admission to intensive care after the procedure. In many patients, WLL only provides temporary symptomatic relief, and once the lungs refill with surfactant, the WLL procedure needs to be repeated.
As there are no approved drug treatments available for PAP, Savara believes there is a high need for a convenient and efficacious medicinal treatment. Savara believes that inhalation of GM-CSF directly into the lungs has the potential to replace the inactivated GM-CSF, and thereby to restore the surfactant clearing activity of the alveolar macrophages. As a result, Savara believes that inhaled GM-CSF has the potential for considerable improvement in oxygenation and exercise tolerance. An injectable form of GM-CSF, sargramostim (Leukine®, Sanofi-Aventis), is approved and on the market in the United States for IV and subcutaneous administration for the treatment of neutropenia caused by cancer chemotherapy, but there is currently no inhalation formulation of GM-CSF available.
The potential benefits of inhaled GM-CSF in PAP, together with the availability of sargramostim, have stimulated independent clinicians and academic researchers in the United States, Europe, and Japan to study the safety and efficacy of GM-CSF, administered by inhalation, in PAP patients. Several such investigator-sponsored open-label clinical studies and case studies of inhaled GM-CSF treatment have been published, with promising results on the efficacy and safety of the treatment.9,10,11 In total, treatment of more than 80 PAP patients with
| 9 | Tazawa R, Trapnell BC, Inoue Y, Arai T, Takada T, Nasuhara Y, et al. Inhaled Granulocyte/Macrophage–Colony Stimulating Factor as Therapy for Pulmonary Alveolar Proteinosis. Am J Resp Crit Care Med 181: 1345-1354, 2010 |
| 10 | Wylam ME, Ten R, Prakash UB, Nadrous HF, Clawson ML and Anderson PM (2006). Aerosol granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis. Eur Respir J 27(3): 585-93 |
| 11 | Papiris SA, Tsirigotis P, Kolilekas L, Papadaki G, Papaioannou AI, Triantafillidou C, et al. (2014). Long-term inhaled granulocyte macrophage-colony-stimulating factor in autoimmune pulmonary alveolar proteinosis: effectiveness, safety, and lowest effective dose. Clin Drug Investig 34(8): 553-64 |
21
Table of Contents
inhaled GM-CSF has been reported in open-label studies or retrospective cohorts, as well as several individual case reports. Whereas the majority of the patients described in the literature received sargramostim, the results indicate that both sargramostim and molgramostim have the potential for a very positive impact on oxygenation and clinical symptoms in PAP patients.
According to Savara’s review of published literature, few safety issues related with molgramostim or sargramostim inhalation in patients with PAP have been reported. However, there is still limited information available on the long-term safety of inhaled GM-CSF. In indications other than PAP, more than 100 patients, mainly with a cancer diagnosis, have received inhaled sargramostim, in doses up to 4000 µg/day. Pulmonary toxicity was the most frequently reported toxicity at high doses. An increase in both number and severity of adverse events with increasing dose has been observed. However, due to the underlying diseases it was often difficult for the investigators to assess causality of the adverse event cases.
Molgradex Product Description
Molgradex is a novel inhaled formulation of recombinant human GM-CSF being developed for the treatment of PAP. The active drug substance, molgramostim, is a non-glycosylated form of GM-CSF. GM-CSF is an endogenous growth factor that stimulates the proliferation and differentiation of hematopoietic cells (blood and immune cells), mainly granulocytic and monocytic cell lines, which serve as the body’s first line of defense against bacteria and viruses, and also function to clear cellular debris and waste substances from the body. Molgramostim is produced in a strain of Escherichia coli bearing a genetically engineered plasmid containing a human GM-CSF gene.
Molgradex, is a sterile nebulizer solution in a vial containing 300 µg of molgramostim, designed to be administered once daily by inhalation via a high efficiency nebulizer (Investigational eFlow Nebuliser System, PARI Pharma GmbH, Germany). The PARI eFlow Nebulizer system for use with investigational drug products is a reusable electronic inhalation system that has been optimized for administration of Molgradex.
Savara anticipates that Molgradex will be used as a long-term therapy in patients with PAP. The optimal duration of treatment is currently not known, and is likely to vary between patients depending on the disease severity and the natural course of their disease. Molgradex treatment may not entirely eliminate the need for WLL in all patients, but based on interviews conducted by Savara, PAP centers that have experimented with long-term inhaled GM-CSF have seen a considerable reduction of WLL procedures.
Molgradex was granted Orphan Drug Designation by the FDA in October, 2012, and by EMA in July, 2013, for the treatment of PAP. Safety and tolerability of inhaled Molgradex has been tested in a Phase 1 clinical study in 42 healthy human volunteers. Safety and efficacy of inhaled Molgradex in PAP patients is currently being tested in a Phase 2/3 clinical study in up to 51 PAP patients. Since 2014, Molgradex has been available in several European countries for the treatment of PAP for named patients following unsolicited physician requests.
Clinical Development of Molgradex
Phase 2/3
Savara is currently conducting a Phase 2/3 clinical study on Molgradex in Europe and Japan in PAP patients. Based on the scientific advice received from the EMA, Savara believes the study has the potential to be accepted as the sole pivotal study in support of a marketing authorization application in the European Union. The aim of this randomized, double-blind, placebo-controlled study is to compare efficacy and safety of Molgradex with placebo in up to 51 PAP patients. In the study, Molgradex 300 µg is administered once daily for up to 24 weeks, with a follow-up period up to 48 weeks.
Patients diagnosed with autoimmune PAP and fulfilling all other entry criteria are randomized to receive double-blind treatment for up to 24 weeks in one of three treatment arms: 1) Molgradex 300 µg administered
22
Table of Contents
once daily, 2) Molgradex 300 µg and matching placebo administered daily in 7-day intermittent cycles of each, or 3) inhaled placebo administered once daily. The study is conducted at multiple sites in the European Union, Russia, Israel and Japan.
The primary endpoint is the absolute change from baseline of arterial-alveolar oxygen gradient ((A-a)DO2) after 24 weeks of treatment. This endpoint is a measure of patient’s oxygenation status, and the endpoint value is expected to decrease as the physical obstacle of gas exchange is reduced by clearance of excess surfactant from the lungs. Key secondary endpoints assessed after 24 weeks of treatment include the number of patients in need of WLL during 24-week treatment, as well as change in the vital capacity of the lungs after 24-week treatment.
Based on the sample size calculation for the study, 42 evaluable patients (14 in each treatment group) are required to be randomized to have 90 % power to detect a difference of 10 mmHg in A-a(DO2) between the two active arms combined and placebo, using a significance level of 0.01. To account for potential study discontinuations or non-evaluable patients, a total of up to 51 patients is planned to be randomized.
A data safety monitoring board, or DSMB, provides safety oversight in the Phase 2/3 study. Following its first meeting in October, 2016, no concerning safety issues were identified and the DSMB endorsed continuation of the study as planned.
Savara has conducted a Type C meeting with the FDA to seek guidance on the nonclinical and clinical requirements for an NDA submission in the United States. The FDA acknowledged that a single Phase 3 study may potentially be sufficient to support approval of Molgradex for treatment of PAP, provided that it demonstrates persuasive evidence of efficacy across clinically meaningful endpoints. Whereas the current study design and sample size of the IMPALA study may not be acceptable to the FDA as a sole pivotal study, the FDA gave initial guidance on modifications of the study that could potentially make it acceptable as the sole study for NDA submission and approval. Savara will diligently continue its interaction with the FDA in order to reach agreement on the clinical program structure and details, and targets to complete the negotiations by the end of the third quarter of 2017. The final outcome may involve the amendment of the IMPALA study to serve as a sole pivotal study, or the conduct of a separate pivotal clinical study prior to submitting an NDA.
Completed Clinical Studies
Phase 1
In a Phase 1 Molgradex study in 42 healthy adult volunteers, the drug was generally well tolerated and produced dose-dependent increases in total and differential white blood cell (WBC) counts consistent with the known pharmacologic effect of GM-CSF. The study was a randomized, double-blind, placebo-controlled, single ascending dose (SAD) and multiple ascending dose (MAD) study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of Molgradex. In the SAD part, 18 subjects were included with four subjects in each of the three SAD dose levels (150 µg, 300 µg and 600 µg) and six subjects received placebo. In the MAD part, 24 subjects were included with nine subjects in each of the two MAD dose levels (300 µg or 600 µg) and six subjects received placebo for six days.
In the SAD part, GM-CSF was absorbed into the systemic circulation with a tmax of two hours after inhalation of Molgradex, however, at picogram levels, 50 to 100 times lower than has been observed after similar doses of GM-CSF administered intravenously. Total systemic exposure (AUClast) increased with dose, ranging between 13 and 138 pg•h/mL and maximum concentration (Cmax) ranging between 9.1 and 41 pg/mL (Cmax was similar for the 300 and 600 µg dose levels). In the MAD part, there was evidence of some accumulation after multiple dosing; Cmax increased from 32 pg/mL on Day 1 to 90 pg/mL on Day 6 at the 300 µg dose, and from 96 pg/mL on Day 1 to 251 pg/mL on Day 6 at the 600 µg dose level. Likewise, AUClast increased from 97 to 248 pg•h/mL from Days 1 to 6 for the 300 µg dose level and from 350 to 802 pg•h/mL for the 600 µg dose level. Minimum measurable plasma concentrations (Cmin) on Day 6 were 3.6 and 5.1 pg/mL measured at 8 and 12 hours, respectively for the 300 and 600 µg dose levels.
23
Table of Contents
In subjects treated with Molgradex, a slight increase in total WBC and differential counts (primarily within normal reference ranges) was observed in a dose-dependent manner, in-line with the known biological mode-of-action of GM-CSF, as illustrated in the graph below.
Mean WBC Over Time and After Multiple Ascending Doses

The primary aim of the Phase 1 study was to assess the safety and tolerability of Molgradex. No meaningful difference in the frequency or severity of AEs was observed between Molgradex 300 µg and placebo. The most common AE was cough, reported in 21 out of 30 (70 %) subjects receiving Molgradex and 8 out of 12 (67 %) patients receiving placebo, and there was no difference in the causality assessment between the treatment arms. A higher number of treatment-related AEs were observed at the 600 µg dose compared to the 300 µg dose and placebo in the MAD part. There were no serious or severe adverse events, dose-limiting toxicity or other remarkable findings of clinical concern in the safety data.
Nonclinical Studies
AeroVanc Inhalation Toxicology Studies
The nonclinical toxicology profile of AeroVanc has been characterized in a series of acute and repeated dose inhalation toxicity studies in rats and dogs, as well as ICH/FDA prescribed safety pharmacology studies involving the cardiovascular, pulmonary, and central nervous systems. In these studies, a gradation of dose levels, including the maximum tolerated dose or the maximum technically achievable dose, were evaluated in both species.
24
Table of Contents
Following 28 days of inhalation exposure, there were no indications of systemic toxicity noted in either the rats or dogs. As expected, there were a number of microscopic changes noted along the respiratory tract and in the lungs that were considered to represent local irritative effects, adaptive changes, and normal physiological responses to the impaction of particles along the respiratory tract and deposition of particles in the lungs. A 28-day recovery period showed complete to partial reversibility of the findings, with no notable difference between the active dose groups and the vehicle control group when compared to the air control. Based on the results of these 28-day studies, the No Observed Adverse Effect Level (NOAEL) was established for both species, and AeroVanc was considered safe for the purpose of conducting the Phase 2 study.
After completion of the Phase 2 clinical study, Savara received guidance from the FDA regarding the necessary toxicology studies to support the planned Phase 3 study and NDA submission. In accordance with the FDA’s guidance, a 91-day inhalation toxicology study was conducted in rats. Savara believes that the NOAEL established in this study supports the proposed Phase 3 study with the intended dose level.
A two-year GLP inhalation carcinogenicity study of AeroVanc in rats is mandated by the FDA prior to submission of an NDA. The purpose of this study is to determine whether lifetime pulmonary exposure to AeroVanc at high doses may result in any gross or microscopic indications of neoplasia in rats. The 91-day inhalation toxicology report and the carcinogenicity study protocol have been evaluated by the FDA Carcinogenicity Assessment Committee (CAC) in a Special Protocol Assessment (SPA) to confirm that the study design and dose levels are adequate to meet scientific and regulatory requirements. The CAC has notified Savara of their feedback, which has been considered in finalizing the protocol. The study will be conducted by a specialized contract research organization that has conducted all prior inhalation toxicology studies of AeroVanc, and has the required capabilities and operating procedures in place.
Molgradex Pharmacology Studies
The pharmacology of GM-CSF in the lungs involves stimulation of alveolar macrophage and neutrophil function to maintain alveolar surfactant homeostasis, alveolar stability, lung function, and lung host defense. For example, pulmonary GM-CSF is required for the terminal differentiation of alveolar macrophages and acquisition of numerous functions including expression of multiple receptors, non-specific and receptor-mediated endocytosis and phagocytosis, for pulmonary neutrophil recruitment during infection, clearance of bacteria, viruses, mycobacteria, and other pathogens, as well as for surfactant clearance.
The pharmacodynamics of human GM-CSF receptor activation by Molgradex was determined as part of Savara’s studies of species evaluation and selection for inhalation toxicology and reproductive toxicology studies. As illustrated below, the effective concentration of molgramostim from Molgradex required to stimulate a half maximal receptor signaling response (EC50), as measured by phosphorylation of STAT5, was similar to that of commercially available rhGM-CSF. Thus, Molgradex is expected to possess the expected biological regulatory action of GM-CSF on alveolar macrophages in the lungs.
25
Table of Contents
GM-CSF receptor function by Molgradex or control recombinant human GM-CSF

Further in vitro or in vivo nonclinical studies investigating the pharmacological activity of Molgradex are not planned.
Molgradex toxicology studies
The nonclinical toxicology profile of Molgradex has been characterized in a series of repeated dose inhalation toxicology studies and safety pharmacology studies in cynomolgus monkeys, as well as reproductive toxicology studies in rabbits. In these studies, a gradation of dose levels was evaluated in the respective species.
Three GLP-compliant inhalation toxicology studies were conducted, including a 6-week inhalation toxicity study in young sexually immature monkeys, a 13-week inhalation toxicity study in sexually mature monkeys used to explore effects on male and female reproductive organs, and a 26-week inhalation toxicity study to investigate chronic toxicity. All studies are fully compliant with relevant guidelines from ICH/FDA.
After inhalation of Molgradex, local effects in the lungs were characterized by infiltrating inflammatory cells, mostly macrophages, accompanied by an increased cellularity in the lymphoid tissue that is associated with the respiratory tract and minimal to mild exudation of red blood cells into the alveoli. The infiltration of inflammatory cells was not associated with any other signs of inflammation or impaired lung function, and it was considered an exaggerated pharmacological effect of molgramostim. The severity of the findings was graded slight at the lowest dose level, and moderate above this level. Duration of treatment did not affect the severity of this finding. Reduced severity of the lung and tracheobronchial changes following a four-week recovery period suggested partial resolution of the changes.
Based on the three studies conducted in monkeys, a NOAEL was established, and Molgradex was considered safe for the purpose of conducting Phase 1 and Phase 2/3 studies, with a safety margin of greater than five-fold using a clinical dose of 300 µg once daily.
Cardiovascular and respiratory parameters and effects on the central nervous system were evaluated in the 6-week and 26-week repeat dose inhalation toxicology studies. It was concluded that repeated daily inhalation of Molgradex does not exert any clinically relevant effects on the heart, the lung or the central nervous system in cynomolgus monkeys.
26
Table of Contents
An embryo-fetal and developmental (EFD) toxicity study with Molgradex was conducted in rabbits, which show a similar pharmacological response as humans or monkeys, although at a lower potency. The EFD study revealed increases in post implantation loss, decreases in the number of live implants, effects on sex ratio and a slight increase in the incidence of major malformations in fetuses at the highest dose (150 µg/kg/day), consistent with findings from other rhGM-CSF products. Studies in sexually mature monkeys have shown that molgramostim has no effect on male and female reproductive organs. Accordingly, appropriate risk minimization strategies are implemented for the clinical studies, and will be implemented for commercial stage use.
In addition to the studies conducted, a pre- and postnatal development study will be conducted prior to NDA submission.
Manufacturing and Supply
Savara does not own or operate manufacturing facilities to produce clinical or commercial quantities of any of its product candidates. Savara has fee-for-service contracts with well-established drug substance manufacturers, as well as drug product manufacturers covering all steps of the manufacturing process of its product candidates, and expects to continue utilizing this outsourcing model in the foreseeable future. All of the vendors used by Savara conduct their operations under current Good Manufacturing Practices, or cGMP, a regulatory standard for the manufacture of pharmaceuticals.
AeroVanc Manufacturing
AeroVanc is a high-performance inhalation powder formulation of vancomycin hydrochloride, applying a commercially-available capsule inhaler. The drug substance used in AeroVanc, Vancomycin Hydrochloride USP, is produced using microbial fermentation followed by purification, and is sourced from Xellia Pharmaceuticals Aps (Copenhagen, Denmark), a commercial manufacturer with two manufacturing facilities, one in China and one in Denmark. Both sites use the same cell line and manufacturing processes, and produce material of comparable quality. A long-term commercial supply agreement has been established with Xellia Pharmaceuticals Aps.
AeroVanc inhalation powder is a spray-dried powder containing a ratio of 9:1 by weight of vancomycin hydrochloride and l-leucine. L-leucine is an essential amino acid and has GRAS status as a food additive. Formulation studies showed that the addition of l-leucine improves inhalation performance in vitro, as measured by improved emitted dose and fine particle dose. The powder manufacturing is carried out by Hovione LLC (East Windsor, NJ), a vendor with two operational sites, one in the United States and one in Europe, with the same base equipment in each facility, that could be upgraded to produce material of comparable quality. The proprietary AeroVanc spray drying process creates very fine particles (smaller than five microns) required for efficient delivery to the lungs. Proprietary nozzle and cyclone technologies were developed to meet product performance and manufacturing throughput requirements. The powder production process has been successfully scaled-up from laboratory to commercial equipment. A long-term commercial supply agreement is under negotiation with Hovione LLC.
The finished product is manufactured from bulk AeroVanc powder by GlaxoSmithKline (GSK, Brentford, UK). At this final part of the manufacturing process, AeroVanc powder is conditioned and automatically filled into capsules each containing 16 mg of vancomycin. The capsules are then packaged into aluminum foil blisters to protect them from light and moisture. A long-term commercial supply agreement has been established with GSK for the finished product.
The inhaler device used for AeroVanc is manufactured by Plastiape S.p.A. (Lecco, Italy). The device was approved in the United States as part of the Aridol® new drug application (NDA 022368) on October 5th, 2010. A cosmetically modified version of the device was approved in the United States as part of the Arcapta® Neohaler® new drug application (NDA 022383) on July 1st, 2011. An exclusive long-term commercial supply agreement has been established with Plastiape S.p.A.
27
Table of Contents
Savara has worked with its manufacturing partners to scale up processes, improve yields and production rates, and to transfer processes to commercial facilities with commercial equipment. Savara is producing the supplies for the pivotal Phase 3 clinical study utilizing the same manufacturing sites, equipment and processes that will be used for commercial supply.
Molgradex Manufacturing
The drug substance in Molgradex, molgramostim, is currently manufactured by Gema Biotech S.A. (GEMA, Buenos Aires, Argentina). All clinical and nonclinical studies to date have used material sourced from GEMA. In 2015, Savara decided to transfer the production to a European manufacturer, Synco Bio Partners B.V. (Synco, Amsterdam, The Netherlands), to secure commercial supply of the drug substance. The technology transfer process from GEMA to Synco is currently ongoing.
The drug product, Molgradex, is currently manufactured at Miltenyi Biotec GmbH (Berglisch Gladbach, Germany). The Molgradex formulation was initially developed to contain several excipients commonly used in freeze-dried formulations used for IV administration. More detailed formulation studies of the inhaled product showed that the physico-chemical stability and potency of the drug product was independent of the presence of these excipients. Accordingly, a simplified formulation without these excipients is in development, and Savara anticipates using this formulation for commercial supply. After the technology transfer process of the drug substance to Synco is complete, manufacture of the drug product will also be carried out at Synco. A master services agreement covering both the drug substance and the drug product has been established with Synco. A long-term commercial supply agreement will be established following the technology transfer.
Molgradex is administered to the lungs using the eFlow Nebulizer System, manufactured by PARI Pharma GmbH (Stamberg, Germany). The eFlow nebulizer has been CE certified (CE 0123) according to the Medical Devices Directive 93/42/EEC (as amended by Directive 2007/47/EC) as a class IIa device. The device has a 510(k) approval in US as a general device. Savara has an exclusive license and a long-term supply agreement with PARI covering the eFlow nebulizer for the administration of recombinant human GM-CSF.
Commercialization
Savara owns exclusive rights to AeroVanc and Molgradex in the United States, and all other major markets, except for Japan, where Savara has licensed the Molgradex rights to Nobelpharma Co., Ltd (Tokyo, Japan). Savara plans to pursue regulatory approvals for its products in the United States and the European Union, and to independently commercialize AeroVanc and Molgradex in the United States. In doing so, Savara may engage with strategic partners to help implement optimal sales and promotion activities. Savara’s commercialization strategy will target key prescribing physicians, as well as provide patients with support programs to ensure product access. Outside of the United States, Savara plans to seek partners to commercialize its products via out-licensing agreements or other similar commercial arrangements.
License and Supply Agreements
Plastiape SpA
In September 2012, Savara entered into a supply agreement related to AeroVanc with Plastiape SpA, which was subsequently amended in June 2016 (the “Plastiape Agreement”). Pursuant to the terms of the Plastiape Agreement, Plastiape will supply dry powder inhalers to Savara on an exclusive basis for use with vancomycin for the diagnosis, management, prevention or treatment of lung diseases. Pricing under the Plastiape Agreement is on a per unit basis, with the per unit price decreasing as the volume increases.
Xellia Pharmaceuticals ApS
In September 2016, Savara entered into a supply agreement related to the supply of the API for AeroVanc with Xellia Pharmaceuticals (the “Xellia Agreement”). Pursuant to the Xellia Agreement, Savara is obligated to
28
Table of Contents
purchase all of its requirements of the API from Xellia. The pricing under the Xellia Agreement is a set price per kg, with the price decreasing upon the commercial launch of AeroVanc.
PARI Pharma GmbH
In November 2014, Serendex entered into a license and collaboration agreement related to Molgradex with PARI Pharma GmbH (the “PARI License Agreement”), which Savara assumed as part of the Serendex Acquisition. Under the PARI License Agreement, Savara has a worldwide, exclusive license to commercialize PARI’s eFlow Technology Nebulizer device for the pulmonary delivery of any liquid formulation containing hGM-CSF as the sole active pharmaceutical ingredient for nebulization. Additionally, Savara has the option to change the device subject to the PARI License Agreement to PARI’s eFlow Technology Nebulizer CS and, until marketing approval, the option to negotiate an extension to the license to cover commercialization of the drug for pulmonary delivery via the PARI eFlow Inline device for the treatment of VAP and/or ARDS.
Under the terms of the PARI License Agreement, Savara is not permitted to work with third parties to develop any inhalation device or nebulizer for the pulmonary delivery of a pharmaceutical product containing hGM-CSF as the sole active ingredient. This restriction extends until (i) in the European Economic Area, marketing approval of the product in Europe or the United States, whichever is later, or (ii) in the rest of the world, the term of the PARI License Agreement.
In consideration of rights granted by PARI, Serendex paid a onetime upfront fee and agreed to pay an hourly rate for work performed by PARI under work orders issued pursuant to the PARI License Agreement. Additionally, Savara is obligated to make future milestone payments to PARI based upon (i) the successful completion of certain clinical trials, (ii) submissions for regulatory approval in the United States, the European Union or Japan, and (iii) the first marketing approval for the product in the United States, the European Union or Japan.
If Savara successfully commercializes any product candidate subject to the PARI License Agreement in a country, Savara is responsible for royalty payments equal to a percentage of net sales. Savara is obligated to make such royalty payments until the later of (i) the expiration of the last valid claim in an issued patent covering a portion of the PARI device in the applicable country or (ii) 15 years after the first commercial sale of Molgradex with the PARI device in that country (the “PARI Royalty Period”). If there is no such valid patent claim covering the applicable PARI device, the royalty owed to PARI will be decreased by a specified percentage.
The license term extends on a country by country basis until the end of the PARI Royalty Period or until mutually agreed by the parties.
In April 2015, Serendex entered into a commercial supply agreement with PARI (the “PARI Supply Agreement”) related to the supply of the PARI eFlow Technology Nebulizer and related accessories for commercial use with its products after marketing approval is obtained. Savara assumed the PARI Supply Agreement as part of the Serendex Acquisition. Pursuant to the terms of the PARI Supply Agreement, Savara is obligated to purchase from PARI (i) within the European Economic Area, (a) during the first five years from marketing approval, all of its requirements for the device and related accessories and (b) thereafter 80% and (ii) in the rest of the world, all of its requirements during the PARI Royalty Period. Pricing is on a per unit basis, with a reduction in price once purchasing volumes reach over 5,000 for devices and starter kits and over 40,000 for nebulizer handsets in a twelve month period.
GEMA Biotech S.A.
In December 2012, Serendex entered into a supply and license agreement related to supplying the API for Molgradex with GEMA Biotech S.A., which was subsequently amended by an addendum in February 2016 (the
29
Table of Contents
“GEMA Agreement”). Savara assumed the GEMA Agreement as part of the Serendex Acquisition. Under the GEMA Agreement, Savara has an exclusive license to market, distribute and sell products based on GEMA recombinant hGM-CSF for any disease to be treated by inhalation, local pulmonary administration, parenteral administration, or local administration of the API in any territory except Latin America, Central America and Mexico. Under the original GEMA Agreement, GEMA is the sole supplier of the API.
As consideration for the rights granted by GEMA, Savara is required to pay GEMA an agreed upon price per vial of 1 gram of the API. Additionally, if Savara successfully develops, registers and obtains approval by the proper health authorities, Savara must pay GEMA a single digit percentage royalty on annual net sales. There is no minimum royalty, and no signing fee or milestones are included in the royalty payments. Additionally, Savara has a commitment to acquire a working cell bank and a master cell bank for $1,950,000 from this API manufacturer in the third quarter of 2017.
Pursuant to the terms of the February 2016 addendum, GEMA granted an exclusive worldwide license to Serendex to transfer the manufacture of the API to Synco Bio Partners B.V., and agreed to sell the master cell bank and working cell bank to Serendex (now Savara). Upon the completion of the purchase by Savara of the master cell bank and working cell bank, the royalty payable to GEMA set forth above decreases.
Government Regulation
The FDA and other regulatory authorities at federal, state, and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and post-approval reporting of drugs, such as those Savara is developing. Savara, along with third-party contractors, will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries in which it wishes to conduct studies or seek approval or licensure of its product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources.
Government Regulation of Drugs
The process required by the FDA before drug product candidates may be marketed in the United States generally involves the following:
| • | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices, or GLP, regulation; |
| • | submission to the FDA of an IND, which must become effective before clinical trials may begin and must be updated annually or when significant changes are made; |
| • | approval by an independent Institutional Review Board, or IRB, or ethics committee for each clinical site before a clinical trial can begin; |
| • | performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed product candidate for its intended purpose; |
| • | preparation of and submission to the FDA of a New Drug Application, or NDA, after completion of all required clinical trials; |
| • | a determination by the FDA within 60 days of its receipt of a NDA to file the application for review; |
| • | satisfactory completion of an FDA Advisory Committee review, if applicable; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with current Good Manufacturing |
30
Table of Contents
| Practices, or cGMP, and to assure that the facilities, methods and controls are adequate to preserve the product’s continued safety, purity and potency, and of selected clinical investigational sites to assess compliance with current Good Clinical Practices, or cGCPs; and |
| • | FDA review and approval of the NDA to permit commercial marketing of the product for particular indications for use in the United States, which must be updated annually and when significant changes are made. |
The testing and approval process requires substantial time, effort and financial resources, and Savara cannot be certain that any approvals for its product candidates will be granted on a timely basis, if at all. Prior to beginning the first clinical trial with a product candidate, Savara must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with cGCPs, which include the requirement that all research subjects provide their informed consent for their participation in any clinical study. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an independent Institutional Review Board, or IRB, for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site, and must monitor the study until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical study sponsor, known as a data safety monitoring board, which provides authorization for whether or not a study may move forward at designated check points based on access to certain data from the study and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical study results to public registries.
For purposes of NDA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
| • | Phase 1. The drug product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, the initial human testing is often conducted in patients. |
| • | Phase 2. The drug product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| • | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency, and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk to benefit ratio of the product and provide an adequate basis for product labeling. |
31
Table of Contents
| • | Phase 4. In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. These so-called Phase 4 studies may be required as a condition to approval of the NDA. |
Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within a specified period, if at all, and there can be no assurance that the data collected will support FDA approval or licensure of the product. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the drug characteristics of the product candidate, and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
NDA Submission and Review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a NDA requesting approval to market the product for one or more indications. The NDA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators. The submission of a NDA requires payment of a substantial User Fee to FDA, and the sponsor of an approved NDA is also subject to annual product and establishment user fees. These fees are typically increased annually. A waiver of user fees may be obtained under certain limited circumstances.
Within 60 days following submission of the application, the FDA reviews a NDA to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any NDA that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the NDA must be resubmitted with the additional information. Once a NDA has been filed, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA reviews a NDA to determine, among other things, whether a product is safe and effective for the indication being pursued, and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety and effectiveness. The FDA may convene an advisory committee to provide clinical insight on application review questions. Before approving a NDA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a NDA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all, and Savara may encounter difficulties or unanticipated costs in its efforts to secure necessary governmental approvals, which could delay or preclude us from marketing its products. After the FDA evaluates a NDA and conducts
32
Table of Contents
inspections of manufacturing facilities where the investigational product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may request additional information or clarification. The FDA may delay or refuse approval of a NDA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
If regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the NDA with a Risk Evaluation and Mitigation Strategy, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance with pre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization, and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of its products under development.
A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of new drugs that meet certain criteria. Specifically, new drug products are eligible for fast track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. For a fast track product, the FDA may consider sections of the NDA for review on a rolling basis before the complete application is submitted if relevant criteria are met. A fast track designated product candidate may also qualify for priority review, under which the FDA sets the target date for FDA action on the NDA at six months after the FDA accepts the application for filing. Priority review is granted when there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If criteria are not met for priority review, the application is subject to the standard FDA review period of 10 months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval.
Under the accelerated approval program, the FDA may approve a NDA on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the biologic’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit. In addition, the Food and Drug Administration Safety and Innovation Act, or FDASIA, which was enacted and signed into law in 2012, established breakthrough therapy designation. A sponsor may seek FDA designation of its product candidate as a breakthrough therapy if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Sponsors may request the FDA to designate a breakthrough therapy at the time of or any time after the submission of an IND, but ideally before an end-of-phase 2 meeting with FDA. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application, which may include holding meetings with the sponsor and the review team throughout the development of the therapy; providing timely advice to, and interactive communication with, the sponsor regarding the development of the drug to ensure that the development program to gather the nonclinical
33
Table of Contents
and clinical data necessary for approval is as efficient as practicable; involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review; assigning a cross-disciplinary project lead for the FDA review team to facilitate an efficient review of the development program and to serve as a scientific liaison between the review team and the sponsor; and considering alternative clinical trial designs when scientifically appropriate, which may result in smaller or more efficient clinical trials that require less time to complete and may minimize the number of patients exposed to a potentially less efficacious treatment. Breakthrough designation also allows the sponsor to file sections of the NDA for review on a rolling basis. Savara may seek designation as a breakthrough therapy for some or all of its product candidates.
Fast Track designation, priority review and breakthrough therapy designation do not change the standards for approval but may expedite the development or approval process.
Orphan Drug Status
Under the Orphan Drug Act, the FDA may grant orphan drug designation to drug candidates intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the United States, or more than 200,000 individuals in the United States and for which there is no reasonable expectation that costs of research and development of the drug for the indication can be recovered by sales of the drug in the United States. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Although there may be some increased communication opportunities, orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a drug candidate that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve any other applications, including a full NDA, to market the same drug for the same indication for seven years, except in very limited circumstances, such as if the second applicant demonstrates the clinical superiority of its product or if FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug for the same disease or condition, or the same drug for a different disease or condition. Among the other benefits of orphan drug designation are tax credits for certain research and a waiver of the NDA application user fee.
Orphan drug exclusivity could block the approval of Savara’s drug candidates for seven years if a competitor obtains approval of the same product as defined by the FDA or if Savara’s drug candidate is determined to be contained within the competitor’s product for the same indication or disease.
As in the United States, designation as an orphan drug for the treatment of a specific indication in the European Union, must be made before the application for marketing authorization is made. Orphan drugs in Europe enjoy economic and marketing benefits, including up to 10 years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan designated product.
The FDA and foreign regulators expect holders of exclusivity for orphan drugs to assure the availability of sufficient quantities of their orphan drugs to meet the needs of patients. Failure to do so could result in the withdrawal of marketing exclusivity for the orphan drug.
GAIN Exclusivity for Antibiotics
In 2012, Congress passed legislation known as the Generating Antibiotic Incentives Now Act, or GAIN Act. This legislation is designed to encourage the development of antibacterial and antifungal drug products that treat
34
Table of Contents
pathogens that cause serious and life-threatening infections. To that end, the new law grants an additional five years of exclusivity upon the approval of an NDA for a drug product designated by FDA as a QIDP. Thus, for a QIDP with Orphan Designation, the periods of five-year exclusivity and seven-year orphan drug exclusivity, would become 12 years.
A QIDP is defined in the GAIN Act to mean “an antibacterial or antifungal drug for human use intended to treat serious or life-threatening infections, including those caused by (1) an antibacterial or antifungal resistant pathogen, including novel or emerging infectious pathogens” or (2) certain “qualifying pathogens.” A “qualifying pathogen” is a pathogen that has the potential to pose a serious threat to public health (such as resistant Gram-positive pathogens, multi-drug resistant Gram-negative bacteria, multi-drug resistant tuberculosis, and C. difficile) and that is included in a list established and maintained by FDA. A drug sponsor may request the FDA to designate its product as a QIDP any time before the submission of an NDA. The FDA must make a QIDP determination within 60 days of the designation request. A product designated as a QIDP will be granted priority review by the FDA and can qualify for “fast track” status.
The additional five years of exclusivity under the GAIN Act for drug products designated by the FDA as QIDPs applies only to a drug that is first approved on or after July 9, 2012. Additionally, the five year exclusivity extension does not apply to: a supplement to an application under FDCA Section 505(b) for any QIDP for which an extension is in effect or has expired; a subsequent application filed with respect to a product approved by the FDA for a change that results in a new indication, route of administration, dosing schedule, dosage form, delivery system, delivery device or strength; or a product that does not meet the definition of a QIDP under Section 505(g) based upon its approved uses.
Post-Approval Requirements
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, distribution, and advertising and promotion of the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual user fee requirements for any marketed products and the establishments at which such products are manufactured, as well as new application fees for supplemental applications with clinical data. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with GMP, which impose certain procedural and documentation requirements upon Savara and its third-party manufacturers. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon us and any third-party manufacturers that Savara may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. Savara cannot be certain that it or its present or future suppliers will be able to comply with the cGMP regulations and other FDA regulatory requirements. If Savara’s present or future suppliers are not able to comply with these requirements, the FDA may, among other things, halt its clinical trials, require them to recall a product from distribution, or withdraw approval of the NDA.
Future FDA and state inspections may identify compliance issues at Savara’s facilities or at the facilities of its contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary, FDA-initiated or judicial action that could delay or prohibit further marketing.
The FDA may withdraw approval of an NDA if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown
35
Table of Contents
problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| • | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| • | fines, warning letters or holds on post-approval clinical studies; |
| • | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
| • | product seizure or detention, or refusal to permit the import or export of products; or |
| • | injunctions or the imposition of civil or criminal penalties. |
The FDA closely regulates the marketing, labeling, advertising and promotion of drugs and biologics. A company can make only those claims relating to safety and efficacy that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by Savara and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturer’s communications on the subject of off-label use of their products.
Government Regulation of Combination Products
Savara’s products under development will be regulated as combination products, which means that they are comprised of two or more different components that, if marketed individually, would be subject to different regulatory paths and would require approval of independent marketing applications by the FDA. A combination product, however, is assigned to a Center with the FDA that will have primary jurisdiction over its regulation on a determination of the combination product’s primary mode of action, which is the single mode of action that provides the most important therapeutic action. Savara believes its product candidates include both a drug and medical device component, and will be regulated as a drug, subject to the review of the FDA’s Center for Drug Evaluation and Research, or CDER, which will have primary jurisdiction over premarket development and approval. FDA’s Center for Devices and Radiological Health, or CDRH, will provide support and review of the inhaler component of the product candidate.
Other Healthcare Laws and Compliance Requirements
Savara’s sales, promotion, medical education, clinical research and other activities following product approval will be subject to regulation by numerous regulatory and law enforcement authorities in the United States in addition to FDA, including potentially the Federal Trade Commission, the Department of Justice, the Centers for Medicare and Medicaid Services, or CMS, other divisions of the U.S. Department of Health and Human Services and state and local governments. Savara’s promotional and scientific/educational programs and interactions with healthcare professionals must comply with the federal Anti-Kickback Statute, the civil False Claims Act, physician payment transparency laws, privacy laws, security laws, and additional federal and state laws similar to the foregoing.
The federal Anti-Kickback Statute prohibits, among other things, the knowing and willing, direct or indirect offer, receipt, solicitation or payment of remuneration in exchange for or to induce the referral of patients,
36
Table of Contents
including the purchase, order or lease of any good, facility, item or service that would be paid for in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. The federal Anti-Kickback Statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on one hand and prescribers, purchasers, formulary managers, and beneficiaries on the other. Although there are a number of statutory exceptions and regulatory safe harbors protecting some common activities from prosecution, the exceptions and safe harbors are drawn narrowly. Practices that involve remuneration that may be alleged to be intended to induce prescribing, purchases or recommendations may be subject to increased scrutiny and review if they do not qualify for an exception or safe harbor. Failure to meet all of the requirements of a particular applicable statutory exception or regulatory safe harbor does not make the conduct per se illegal under the federal Anti-Kickback Statute. Instead, the legality of the arrangement will be evaluated on a case-by-case basis based on a cumulative review of all its facts and circumstances. Several courts have interpreted the statute’s intent requirement to mean that if any one purpose of an arrangement involving remuneration is to induce referrals of federal healthcare covered business, the federal Anti-Kickback Statute has been violated. The government has enforced the federal Anti-Kickback Statute to reach large settlements with healthcare companies based on sham research or consulting and other financial arrangements with physicians. Further, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the False Claims Act. Many states have similar laws that apply to their state health care programs as well as private payers.
Federal false claims and false statement laws, including the federal civil False Claims Act, or FCA, imposes liability on persons and/or entities that, among other things, knowingly present or cause to be presented claims that are false or fraudulent or not provided as claimed for payment or approval by a federal health care program. The FCA has been used to prosecute persons or entities that “cause” the submission of claims for payment that are inaccurate or fraudulent, by, for example, providing inaccurate billing or coding information to customers, promoting a product off-label, submitting claims for services not provided as claimed, or submitting claims for services that were provided but not medically necessary. Actions under the FCA may be brought by the Attorney General or as a qui tam action by a private individual, or whistleblower, in the name of the government. Violations of the FCA can result in significant monetary penalties and treble damages. The federal government is using the FCA, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other illegal sales and marketing practices. The government has obtained multi-million and multi-billion dollar settlements under the FCA in addition to individual criminal convictions under applicable criminal statutes. In addition, certain companies that were found to be in violation of the FCA have been forced to implement extensive corrective action plans, and have often become subject to consent decrees or corporate integrity agreements, restricting the manner in which they conduct their business.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payers; knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services; and willfully obstructing a criminal investigation of a healthcare offense. Like the federal Anti-Kickback Statute, the Affordable Care Act amended the intent standard for certain healthcare fraud statutes under HIPAA such that a person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it in order to have committed a violation.
Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws. Also, many states have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payer, in addition to items and services reimbursed under
37
Table of Contents
Medicaid and other state programs. Additionally, to the extent that Savara’s products, once commercialized, are sold in a foreign country, Savara may be subject to similar foreign laws.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, among other things, imposed new reporting requirements on certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, for payments or other transfers of value made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Covered manufacturers are required to collect and report detailed payment data and submit legal attestation to the accuracy of such data to the government each year. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests that are not timely, accurately and completely reported in an annual submission. Additionally, entities that do not comply with mandatory reporting requirements may be subject to a corporate integrity agreement. Certain states also mandate implementation of commercial compliance programs, impose restrictions on covered manufacturers’ marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians and other healthcare professionals.
Savara may also be subject to data privacy and security regulation by both the federal government and the states in which it conducts its business. HIPAA, as amended by the Health Information Technology and Clinical Health Act, or HITECH, and their respective implementing regulations, imposes specified requirements on certain health care providers, plans and clearinghouses (collectively, “covered entities”) and their “business associates,” relating to the privacy, security and transmission of individually identifiable health information. Among other things, HITECH makes HIPAA’s security standards directly applicable to “business associates,” defined as independent contractors or agents of covered entities that create, receive, maintain or transmit protected health information in connection with providing a service for or on behalf of a covered entity. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce HIPAA and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, certain states have their own laws that govern the privacy and security of health information in certain circumstances, many of which differ from each other and/or HIPAA in significant ways and may not have the same effect, thus complicating compliance efforts.
If Savara’s operations are found to be in violation of any of such laws or any other governmental regulations that apply to them, Savara may be subject to penalties, including, without limitation, civil and criminal penalties, damages, fines, disgorgement, the curtailment or restructuring of its operations, exclusion from participation in federal and state healthcare programs, imprisonment, contractual damages, reputational harm, and diminished profits and future earnings, any of which could adversely affect its ability to operate its business and its financial results.
In addition to the foregoing health care laws, Savara is also subject to the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws, which generally prohibit companies and their intermediaries from making improper payments to government officials or private-sector recipients for the purpose of obtaining or retaining business. Savara has plans to adopt an anti-corruption policy, which will become effective upon the completion of this offering, and expect to prepare and implement procedures to ensure compliance with such policy. The anti-corruption policy mandates compliance with the FCPA and similar anti-bribery laws applicable to its business throughout the world. However, Savara cannot assure you that such a policy or procedures implemented to enforce such a policy will protect them from intentional, reckless or negligent acts committed by its employees, distributors, partners, collaborators or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties or prosecution and have a negative impact on its business, results of operations and reputation.
38
Table of Contents
Coverage and Reimbursement
Sales of pharmaceutical products depend significantly on the extent to which coverage and adequate reimbursement are provided by third-party payers. Third-party payers include state and federal government health care programs, managed care providers, private health insurers and other organizations. Although Savara currently believes that third-party payers will provide coverage and reimbursement for its product candidates, if approved, Savara cannot be certain of this. Third-party payers are increasingly challenging the price, examining the cost-effectiveness, and reducing reimbursement for medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. The U.S. government, state legislatures and foreign governments have continued implementing cost containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Adoption of price controls and cost containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit Savara’s net revenue and results. Savara may need to conduct expensive clinical studies to demonstrate the comparative cost-effectiveness of its products. The product candidates that Savara develops may not be considered cost-effective and thus may not be covered or sufficiently reimbursed. It is time consuming and expensive for them to seek coverage and reimbursement from third-party payers, as each payer will make its own determination as to whether to cover a product and at what level of reimbursement. Thus, one payer’s decision to provide coverage and adequate reimbursement for a product does not assure that another payer will provide coverage or that the reimbursement levels will be adequate. Moreover, a payer’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Reimbursement may not be available or sufficient to allow them to sell its products on a competitive and profitable basis.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could materially affect Savara’s ability to sell its products profitably. Among policy makers and payers in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
By way of example, in March 2010, the Affordable Care Act was signed into law, intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for the healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Among the provisions of the Affordable Care Act of importance to Savara’s potential drug candidates are:
| • | an annual, nondeductible fee on any entity that manufactures or imports specified branded prescription drugs and biologic agents, apportioned among these entities according to their market share in certain government healthcare programs; |
| • | an increase in the statutory minimum rebates a manufacturer must pay under the Medicaid Drug Rebate Program to 23.1% and 13.0% of the average manufacturer price for branded and generic drugs, respectively; |
| • | a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected; |
| • | a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50% point-of-sale discounts off negotiated prices of applicable brand drugs to eligible beneficiaries during their coverage gap period, as a condition for a manufacturer’s outpatient drugs to be covered under Medicare Part D; |
| • | extension of a manufacturer’s Medicaid rebate liability to covered drugs dispensed to individuals who are enrolled in Medicaid managed care organizations; |
39
Table of Contents
| • | expansion of eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for certain individuals with income at or below 133% of the federal poverty level, thereby potentially increasing a manufacturer’s Medicaid rebate liability; |
| • | expansion of the entities eligible for discounts under the Public Health Service pharmaceutical pricing program; and |
| • | a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research. |
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. These changes include, among others, the Budget Control Act of 2011, which mandates aggregate reductions to Medicare payments to providers of up to 2% per fiscal year effective April 1, 2013, and, due to subsequent legislative amendments, will remain in effect through 2024 unless additional Congressional action is taken. In January 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, which, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws may result in additional reductions in Medicare and other healthcare funding, which could have a material adverse effect on customers for Savara’s product candidates, if approved, and, accordingly, its financial operations.
Savara expects that healthcare reform measures that may be adopted in the future, including the possible repeal and replacement of the Affordable Care Act which the Trump administration has stated is a priority, are unpredictable, and the potential impact on Savara’s operations and financial position are uncertain, but may result in more rigorous coverage criteria and lower reimbursement, and place additional downward pressure on the price that it receives for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent Savara from being able to generate revenue, attain profitability or commercialize their drugs.
Foreign Regulation
In addition to regulations in the United States, Savara will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of its products to the extent Savara chooses to develop or sell any products outside of the United States. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Intellectual Property
Savara strives to protect the proprietary technology that Savara believes is important to its business, including its product candidates and its processes. Savara seeks patent protection in the United States and internationally for its products, their methods of use and processes of manufacture and any other technology to which Savara has rights, as appropriate. Savara also relies on trade secrets that may be important to the development of its business.
Savara owns six issued patents and additional pending patent applications worldwide for a proprietary formulation of AeroVanc. The patents and pending applications are derived from a PCT application (Pub. No. WO2012159103) entitled “Dry Powder Vancomycin Compositions and Associated Methods.” As of March 1, 2017, patents have issued in the United States, Australia, China, Japan, New Zealand, and Singapore.
40
Table of Contents
While Savara does not have any issued patents or pending applications covering Molgradex or its use in pulmonary alveolar proteinosis (PAP), Savara does own a family of issued patents and pending applications derived from a PCT application (Pub. No. WO2008052567) entitled “Enhancing Pulmonary Host Defense via Administration of Granulocyte-Macrophage Colony-Stimulating Factor” covering inhaled GM-CSF for treatment of bacterial, mycobacterial (including Mycobacterium tuberculosis and non-tuberculous Mycobacterium), yeast, and virus infections in the lungs. Patents have been granted in Japan, Australia, and Mexico. Patent applications are currently pending in several other countries, including the United States. An application is also pending in the European Union, where an allowance has been indicated.
Savara’s success will in part depend on the ability to obtain and maintain patent and other proprietary rights in commercially important technology, inventions and know-how related to its business, the validity and enforceability of its patents, the continued confidentiality of its trade secrets as well as its ability to operate without infringing the valid and enforceable patents and proprietary rights of third parties. Savara also relies on continuing technological innovation and in-licensing opportunities to develop and maintain its proprietary position.
Savara cannot be sure that patents will be granted with respect to any of its pending patent applications or with respect to any patent applications it may own or license in the future, nor can Savara be sure that any of its existing patents or any patents it may own or license in the future will be useful in protecting its technology and products. For this and more comprehensive risks related to Savara’s intellectual property, please see “Risk Factors — Risks Related to Savara’s Intellectual Property.”
Trade Secrets
In addition to patents, Savara relies on trade secrets and know-how to develop and maintain its competitive position. For example, significant aspects of Savara’s processes and proprietary technology portfolio are based on unpatented trade secrets and know-how. Trade secrets and know-how can be difficult to protect. Savara seeks to protect its proprietary technology and processes, in part, by confidentiality agreements and invention assignment agreements with its employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect the proprietary information and, in the case of the invention assignment agreements, to grant the company ownership of technologies that are developed through a relationship with a third party. While Savara has confidence in its key individuals, consultants, partner organizations and systems, agreements or security measures may be breached, and there may not be adequate remedies for any breach. In addition, Savara’s trade secrets may otherwise become known or be independently discovered by competitors. To the extent that Savara’s contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Competition
The pharmaceutical industry is highly competitive and subject to continuous technological change. Savara’s potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical and generic drug companies, academic institutions, government agencies and research institutions. Savara believes that key competitive factors affecting the commercial success of its product candidates will be efficacy, safety and tolerability profile, reliability, convenience of dosing, price and reimbursement. Many of Savara’s potential competitors, either alone or with their collaboration partners have substantially greater financial, technical and human resources than Savara, and significantly greater experience in the discovery and development of product candidates, manufacturing, obtaining FDA and other regulatory approvals of products and the commercialization of those products. Accordingly, Savara’s competitors may be faster and more successful in obtaining FDA approval for therapies and achieving widespread market acceptance. Mergers and acquisitions in the pharmaceutical and biotechnology industry may result in even more resources being concentrated among a smaller number of very capable competitors. Savara anticipates facing intense and increasing competition as new drugs enter the market and advanced technologies become available. Savara’s competitors’ products may be
41
Table of Contents
more effective, or more effectively marketed and sold, than any product candidate Savara may commercialize and may render Savara’s therapies obsolete or non-competitive before Savara can recover development and commercialization expenses.
Savara is not aware of any other companies developing inhaled forms of vancomycin. There are several inhaled antibiotics on the market or in development, but Savara is not aware of any other inhaled antibiotic product that would be specifically developed for the treatment of MRSA infection. Certain inhaled antibiotics in development, including levofloxacin, and ciprofloxacin inhalation formulations, may possess some level of in vitro or in vivo activity against MRSA, even though the compounds are not generally considered MRSA-antibiotics. It is therefore possible that such products, if approved, may present a competitive threat to AeroVanc. A combination product containing fosfomycin and tobramycin for inhalation (FTI) was developed by Gilead Sciences (Foster City, CA), and shown in a Phase 2 study to possess activity against Gram-negative and Gram-positive bacteria, including MRSA. Gilead terminated the development of the product, and licensed it to CURx Pharmaceuticals (San Diego, CA) in February, 2014. No clinical studies on FTI have been initiated by CURx. If FTI is developed, and approved, for the treatment of MRSA lung infection in CF, Savara believes it has the potential to present a competitive threat to the commercial success of AeroVanc.
Many small and large pharmaceutical companies have intravenously or orally administered MRSA-antibiotics on the market, and/or in development. Whereas such antibiotics are important in the treatment of many acute and chronic MRSA-infections, such as skin and soft tissue infections, pneumonia, or endocarditis, Savara does not believe these products are practical or sufficiently efficacious and/or safe for long-term management of chronic MRSA lung infection in CF patients. Therefore, Savara does not believe these products and product candidates are a material competitive threat to the commercial success of AeroVanc.
Savara is not aware of any other companies developing an inhaled form of GM-CSF. A glycosylated GM-CSF product, sargramostim (Leukine, Sanofi), is available on the market in the United States, intended for IV or subcutaneous delivery in patients with neutropenia following cancer chemotherapy. Leukine has not been approved, and according to Savara’s knowledge, is not being developed for the treatment of PAP or any other acute or chronic lung disease. The drug substance in Leukine, sargramostim, has been used in a nonclinical research project conducted by NIH/TRND in collaboration with the University of Cincinnati College of Medicine on the potential application of inhaled GM-CSF as a treatment for PAP. No clinical studies have been conducted to date under this collaboration project. Savara is aware of a multicenter clinical study of inhaled Leukine, using a standard commercially available nebulizer, which is currently ongoing in Japan, conducted by a consortium of independent clinical investigators. It is not known to Savara if this study, together with other possibly available related clinical or nonclinical information, may be, or will be, used to support a potential new product approval in Japan. If such a new product would be approved and launched in Japan, Savara believes it has the potential to present a material competitive threat to the commercial success of Molgradex in Japan.
Asset Purchase Agreement with Serendex A/S
On May 13, 2016, Savara entered into a Business Transfer Agreement with Serendex A/S (subsequently named to Serenova) under which Serendex agreed to sell, transfer and assign to Savara all of its assets and subsidiaries, certain of its contracts, and certain of its employees and liabilities (“Serendex Acquisition”). Serendex was a limited liability company incorporated in Denmark and was listed on the Oslo Stock Exchange until May 4, 2016. On July 15, 2016, Savara completed the Serendex Acquisition through its wholly-owned subsidiary, Savara ApS, a limited liability company established under the laws of Denmark.
The Serendex Acquisition was an important step in fulfilling Savara’s vision to become a specialty pharmaceutical company focused on rare respiratory diseases. Serendex was a biopharmaceutical development company advancing a pipeline and portfolio of novel inhalation therapies for the treatment of severe pulmonary conditions. Through the Serendex Acquisition, Savara gained access to the late-stage Molgradex program for the
42
Table of Contents
treatment of PAP, with a Phase 2/3 clinical study (IMPALA study) ongoing in EU and Japan. In addition to Molgradex, Savara gained access to an experienced development team familiar with all aspects of the Molgradex program.
As the purchase consideration, Savara agreed to provide the seller with 3,353,925 shares of Savara’s common stock. In addition to these purchase consideration shares, Savara agreed to pay the seller (i) $5,000,000 upon receipt of marketing approval of Molgradex for the treatment of PAP (the Product) by the European Medicines Agency, (ii) $15,000,000 upon receipt of marketing approval of the Product by the United States Food and Drug Administration, and (iii) $1,500,000 upon receipt of marketing approval of the Product by the Japanese Pharmaceuticals and Medical Devices Agency (the Contingent Milestone Payments).
Employees
As of March 6, 2017, Savara had 15 full-time employees and one part time employee, as well as several full-time or part time consultants. None of Savara’s employees are represented by a labor union or covered by a collective bargaining agreement. Savara considers its relationship with its employees to be good.
Facilities
Savara’s corporate headquarters is located in Austin, Texas, where the company leases approximately 2,800 square feet of office space pursuant to a lease that expires in 2019.
Savara believes that its existing facilities are adequate for its near-term needs. When the lease expires, Savara may look for alternate space for its operations. Savara believes that suitable alternative space would be available if required in the future on commercially reasonable terms.
Legal Proceedings
Savara is not currently a party to any material legal proceedings.
43
Table of Contents
Risks Related to Savara’s Capital Requirements and Financial Condition
Savara has a limited operating history and has incurred significant losses since inception, and expects that it will continue to incur losses for the foreseeable future, which makes it difficult to assess Savara’s future viability.
Savara is a clinical development-stage biopharmaceutical company with a limited operating history upon which to evaluate its business and prospects. Savara has not been profitable since it commenced operations in 2008, and may not achieve profitability. In addition, Savara has limited history as an organization and has not yet demonstrated an ability to successfully overcome many of the risks and uncertainties frequently encountered by companies in new and rapidly evolving fields, particularly in the biopharmaceutical industry. Drug development is a highly speculative undertaking and involves a substantial degree of risk. To date, Savara has not obtained any regulatory approvals for any of its product candidates, commercialized any of its product candidates or generated any product revenue. Savara has devoted significant resources to research and development and other expenses related to its ongoing clinical trials and operations, in addition to acquiring product candidates.
For the year ended December 31, 2016, Savara incurred losses from operations of $10.9 million, and net cash used in operating activities was $8.4 million. At December 31, 2016, Savara had an accumulated deficit of $38.4 million, its cash, cash equivalents and investment securities were $13.4 million, and its working capital was $11.2 million. Savara expects to continue to incur substantial operating losses for the next several years as it advances its product candidates through clinical development, global regulatory approvals, and commercialization. No revenue from operations will likely be available until, and unless, one of its product candidates is approved by the FDA or another regulatory agency and successfully marketed, or Savara enters into an arrangement that provides for licensing revenue or other partnering-related funding, outcomes which Savara may not achieve.
Savara will require substantial additional financing to obtain regulatory approval for AeroVanc and Molgradex, and a failure to obtain this necessary capital when needed on acceptable terms, or at all, could force Savara to delay, limit, reduce or terminate Savara’s product development efforts or other operations.
Since inception, most of Savara’s resources have been dedicated to the development and acquisition of its product candidates, AeroVanc and Molgradex. Savara believes that its existing capital resources will be sufficient to fund its operations for up to 12 months. Savara may raise additional capital from its existing investors prior to the closing of the Merger and may raise additional capital from new investors following the closing of the Merger. Savara will require significant additional capital to continue operations and execute on its current business strategy to develop AeroVanc and Molgradex through to regulatory approval. Savara cannot
44
Table of Contents
estimate with reasonable certainty the actual amounts necessary to successfully complete the development and commercialization of its product candidates and there is no certainty that Savara will be able to raise the necessary capital on reasonable terms or at all.
Savara’s capital requirements for the foreseeable future will depend in large part on, and could increase significantly as a result of, its expenditures on its development programs. Future expenditures on its development programs are subject to many uncertainties, and will depend on, and could increase significantly as a result of, many factors, including:
| • | the number, size, complexity, results and timing of its drug development programs; |
| • | the timing and terms of any collaborative or other strategic arrangement that Savara may establish; |
| • | the number of clinical and nonclinical studies necessary to demonstrate acceptable evidence of the safety and efficacy of its product candidates; |
| • | changes in standards of care which could increase the size and complexity of clinical studies; |
| • | the number of patients who participate, the rate of enrollment, and the ratio of randomized to evaluable patients in each clinical study; |
| • | the ability to locate patients to participate in a study given the limited number of patients available for orphan or ultra-orphan indications; |
| • | the number and location of sites and the rate of site initiation in each study; |
| • | the duration of patient treatment and follow-up; |
| • | the potential for additional safety monitoring or other post-marketing studies that may be requested by regulatory agencies; |
| • | the time and cost to manufacture clinical trial material and commercial product, including process development and scale-up activities, and to conduct stability studies, which can last several years; |
| • | the degree of difficulty and cost involved in securing alternate manufacturers or suppliers of drug product, components or delivery devices, as necessary to meet FDA requirements and/or commercial demand; |
| • | the costs, requirements, timing of, and the ability to, secure regulatory approvals; |
| • | the extent to which Savara increases its workforce and the costs involved in recruiting, training and incentivizing new employees; |
| • | the costs related to developing, acquiring and/or contracting for sales, marketing and distribution capabilities, supply chain management capabilities, and regulatory compliance capabilities, if Savara obtains regulatory approval for a product candidate and commercializes it without a partner; |
| • | the costs involved in evaluating competing technologies and market developments or the loss in sales in case of such competition; and |
| • | the costs involved in establishing, enforcing or defending patent claims and other proprietary rights. |
Additional capital may not be available when Savara needs it, on terms that are acceptable to it or at all. If adequate funds are not available to Savara on a timely basis, it will be required to delay, limit, reduce or terminate its establishment of sales and marketing, manufacturing or distribution capabilities, development activities or other activities that may be necessary to commercialize its product candidates, conduct preclinical or clinical studies, or other development activities.
If Savara raises additional capital through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, it may have to relinquish certain valuable rights to
45
Table of Contents
its product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable. If Savara raises additional capital through public or private equity offerings, the ownership interest of its stockholders will be diluted and the terms of any new equity securities may have preferential rights over its common stock. If Savara raises additional capital through debt financing, it may be subject to covenants limiting or restricting its ability to take specific actions, such as incurring additional debt or making capital expenditures, or subject to specified financial ratios, any of which could restrict its ability to develop and commercialize its product candidates or operate as a business.
Risks Related to Savara’s Business Strategy and Operations
Savara is substantially dependent upon the clinical, regulatory and commercial success of its two product candidates, AeroVanc and Molgradex. Clinical drug development involves a lengthy and expensive process with an uncertain outcome, results of earlier studies and trials may not be predictive of future trial results, and Savara’s clinical trials may fail to adequately demonstrate to the satisfaction of regulatory authorities the safety and efficacy of its two product candidates.
The success of Savara’s business is dependent on its ability to advance the clinical development of AeroVanc for the treatment of persistent methicillin-resistant Staphylococcus aureus (MRSA) infections in the lungs of cystic fibrosis patients and Molgradex for the treatment of patients with pulmonary alveolar proteinosis (PAP). The AeroVanc Phase 3 study is scheduled to start in the United States and Canada in Q3 2017 and the Molgradex Phase 2/3 clinical study (IMPALA) is ongoing in Europe and Japan. Savara expects to announce top-line results from the Phase 2/3 study of Molgradex in the first quarter of 2018.
Clinical testing is expensive and can take many years to complete, and its outcome is inherently uncertain. A failure of one or more of Savara’s clinical trials can occur at any time during the clinical trial process. The results of preclinical studies and early clinical trials of Savara’s product candidates may not be predictive of the results of later-stage clinical trials. There is a high failure rate for drugs proceeding through clinical trials, and product candidates in later stages of clinical trials may fail to show the required safety and efficacy despite having progressed through preclinical studies and initial clinical trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials due to lack of efficacy or adverse safety profiles, notwithstanding promising results in earlier clinical trials, and Savara cannot be certain that it will not face similar setbacks. Even if Savara’s clinical trials are completed, the results may not be sufficient to obtain regulatory approval for its product candidates.
Given the development nature of Savara’s product candidates, Savara is subject to risks associated with initiating, completing and achieving positive outcomes from its current and future clinical trials, including:
| • | slow implementation, enrollment and completion of the clinical trials; |
| • | inability to enroll enough patients in the clinical trials; |
| • | low patient compliance and adherence to dosing and reporting requirements, for example incomplete reporting of patient reported outcomes in the clinical trials or missed doses; |
| • | lack of safety and efficacy in the clinical trials; |
| • | delays in manufacture of supplies for both drug and device components due to delays in formulation, process development, or manufacturing activities; |
| • | requirements for additional nonclinical or clinical studies based on changes to formulation and/or changes to regulatory requirements; |
| • | requirements for additional clinical studies based on inconclusive clinical results or changes in market, standard of care, and/or regulatory requirements; |
46
Table of Contents
If Savara successfully completes the necessary clinical trials for its product candidates, its success will be subject to the risks associated with obtaining regulatory approvals, product launch, and commercialization, including:
| • | FDA rejection of Savara’s NDA submissions for its product candidates; |
| • | regulatory rejection in the EU, Japan, and other markets; |
| • | delays during regulatory review and/or requirements for additional CMC, nonclinical, or clinical studies, resulting in increased costs and/or delays in marketing approval and subsequent commercialization of the product candidates in the United States and other markets; |
| • | inability to consistently manufacture commercial supplies of drug and delivery devices resulting in slowed market development and lower revenue; |
| • | poor commercial sales due to: |
| • | the ability of Savara’s future sales organization or its potential commercialization partners to effectively sell the product candidates; |
| • | Savara’s lack of success in educating physicians and patients about the benefits, administration and use of its product candidates; |
| • | the availability, perceived advantages, relative cost, relative safety and relative efficacy of other products or treatments for the targeted indications of the product candidates; |
| • | low patient demand for the product candidates; |
| • | poor prescription coverage and inadequate reimbursement for its product candidates; |
| • | Savara’s inability to enforce its intellectual property rights in and to its product candidates; and |
| • | reduction in the safety profile of its product candidates following approval. |
Many of these clinical, regulatory and commercial matters are beyond Savara’s control and are subject to other risks described elsewhere in this “Risk Factors” section. Accordingly, Savara cannot assure that it will be able to advance its product candidates further through final clinical development, or obtain regulatory approval of, commercialize or generate significant revenue from them. If Savara cannot do so, or are significantly delayed in doing so, its business will be materially harmed.
If Savara fails to attract and retain senior management and key scientific personnel, it may be unable to successfully develop and commercialize its product candidates.
Savara has historically operated with a limited number of employees that manage third-parties for most development activities. Institutional knowledge is concentrated within a small number of employees. Savara’s success depends in part on its continued ability to attract, retain and motivate highly qualified management, clinical and scientific personnel. Savara’s future success is highly dependent upon the contributions of its senior management, as well as its senior scientists and other members of its senior management team. The loss of services of any of these individuals, who all have at-will employment arrangements with Savara, could delay or prevent the successful development of its product pipeline, completion of its planned clinical trials or the commercialization of its product candidates.
Replacing key employees may be a difficult, costly and protracted process, and Savara may not have other personnel with the capacity to assume all the responsibilities of a key employee upon his/her departure. Transition periods can be difficult to manage and may cause disruption to its business. In addition, there may be intense competition from other companies and organizations for qualified personnel. Other companies and organizations with which Savara competes for personnel may have greater financial and other resources and different risk profiles than Savara, and a history of successful development and commercialization of its product candidates. If Savara cannot attract and retain skilled personnel, as needed, Savara may not achieve its development and other goals.
47
Table of Contents
In addition, the success of Savara’s business will depend on its ability to develop and maintain relationships with respected service providers and industry-leading consultants and advisers. If Savara cannot develop and maintain such relationships, as needed, the rate and success at which Savara can develop and commercialize product candidates may be limited. In addition, its outsourcing strategy, which has included engaging consultants that spend considerable time in its office to manage key functional areas, may subject Savara to scrutiny under labor laws and regulations, which may divert management time and attention and have an adverse effect on its business and financial condition.
Savara does not have, and does not have plans to establish manufacturing facilities. Savara completely relies on third parties for the manufacture and supply of its clinical trial drug and delivery device supplies and, if approved, commercial product materials. The loss of any of these vendors or a vendor’s failure to provide Savara with an adequate supply of clinical trial or commercial product material in a timely manner and on commercially acceptable terms, or at all, could harm its business.
Savara outsources the manufacture of its product candidates and does not plan to establish its own manufacturing facilities. To manufacture Savara’s product candidates, Savara has made numerous custom modifications at CMOs, making Savara highly dependent on these CMOs. For clinical and commercial supplies, if approved, Savara has supply agreements with third party CMOs for drug substance, finished drug product, drug delivery devices and other necessary components of its product candidates. While Savara has secured long-term commercial supply agreements with many of the third party CMOs, Savara would need to negotiate agreements for commercial supply with several important CMOs, and Savara may not be able to reach agreement on acceptable terms. In addition, Savara relies on these third parties to conduct or assist Savara in key manufacturing development activities, including qualification of equipment, developing and validating methods, defining critical process parameters, releasing component materials and conducting stability testing, among other things. If these third parties are unable to perform their tasks successfully in a timely manner, whether for technical, financial or other reasons, Savara may be unable to secure clinical trial material, or commercial supply material if approved, which likely would delay the initiation, conduct or completion of its clinical studies or prevent Savara from having enough commercial supply material for sale, which would have a material and adverse effect on its business.
All manufacturers of Savara’s clinical trial material and, if approved, commercial product, including drug substance manufacturers, must comply with cGMP requirements enforced by the FDA through its facilities inspection program and applicable requirements of foreign regulatory authorities. These requirements include quality control, quality assurance and the maintenance of records and documentation. Manufacturers of Savara’s clinical trial material may be unable to comply with these cGMP requirements and with other FDA, state and foreign regulatory requirements. While Savara or its representatives generally monitor and audit its manufacturers’ systems, Savara does not have full control over their ongoing compliance with these regulations. And while the responsibility to maintain cGMP compliance is shared between Savara and the third-party manufacturer, Savara bears ultimately responsibility for its supply chain and compliance with regulatory standards. Failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay or failure to obtain product approval, product seizure or recall, or withdrawal of product approval.
Currently, Savara does not have alternative vendors to back up its primary vendors of clinical trial material or, if approved, commercial supply material. Identification of and discussions with other vendors may be protracted and/or unsuccessful, or these new vendors may be unsuccessful in producing the same results as the current primary vendors producing the material. Therefore, if its primary vendors become unable or unwilling to perform their required activities, Savara could experience protracted delays or interruptions in the supply of clinical trial material and, ultimately, product for commercial sale, which would materially and adversely affect its development programs, commercial activities, operating results and financial condition. In addition, the FDA or regulatory authorities outside of the United States may require that Savara have an alternate manufacturer of a drug product before approving it for marketing and sale in the United States or abroad and securing such
48
Table of Contents
alternate manufacturer before approval of an NDA could result in considerable additional time and cost prior to NDA approval.
Any new manufacturer or supplier of finished drug product or its component materials, including drug substance and delivery devices, would be required to qualify under applicable regulatory requirements and would need to have sufficient rights under applicable intellectual property laws to the method of manufacturing of such product or ingredients required by Savara. The FDA or foreign regulatory agency may require Savara to conduct additional clinical studies, collect stability data and provide additional information concerning any new supplier, or change in a validated manufacturing process, including scaling-up production, before Savara could distribute products from that manufacturer or supplier or revised process. For example, if Savara were to engage a third party other than its current CMOs to supply the drug substance or drug product for future clinical trial, or commercial product, the FDA or regulatory authorities outside of the United States may require Savara to conduct additional clinical and nonclinical studies to ensure comparability of the drug substance or drug product manufactured by its current CMOs to that manufactured by the new supplier. Changing of suppliers or equipment is particularly challenging for companies like Savara, with inhalation products, because any change could alter the drug product of its performance. The manufacturing of the drug substance of Molgradex, molgramostim, a biological drug substance, as well as the drug product, Molgradex, is currently being transferred to a new manufacturing site. Producing a pharmaceutically and biologically similar product may prove to be challenging, and may take more time and resources that currently anticipated. The transfer of the manufacturing to the new site may also cause regulatory agencies, including the FDA, to require additional nonclinical or clinical studies, which may cause delay or failure to obtain regulatory approval, and incur substantial additional cost.
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling-up initial production. These problems include difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing, and shortages of qualified personnel. Some of Savara’s product candidates have not been manufactured at the scale Savara believes will be necessary to maximize its commercial value and, accordingly, Savara may encounter difficulties in attempting to scale-up production and may not succeed in that effort on a timely basis or at all. In addition, the FDA or other regulatory authorities may impose additional requirements as Savara scales-up initial production capabilities, which may delay its scale-up activities and/or add expense.
If Savara’s manufacturers encounter any of the aforementioned difficulties or otherwise fail to comply with their contractual obligations or there are delays entering commercial supply agreements due to capital constraints, Savara may have insufficient quantities of material to support ongoing and/or planned clinical studies or to meet commercial demand, if approved. In addition, any delay or interruption in the supply of materials necessary or useful to manufacture its product candidates could delay the completion of its clinical studies, increase the costs associated with its development programs and, depending upon the period of delay, require Savara to commence new clinical studies at significant additional expense or terminate the studies completely. Delays or interruptions in the supply of commercial product could result in increased cost of goods sold and lost sales. Savara cannot provide assurance that manufacturing or quality control problems will not arise in connection with the manufacture of its clinical trial material or commercial product, if approved, or that third-party manufacturers will be able to maintain the necessary governmental licenses and approvals to continue manufacturing such clinical trial material or commercial product, as applicable. In addition, AeroVanc and Molgradex are currently manufactured entirely or partially outside the United States and, as a result, Savara may experience interruptions in supply due to shipping or customs difficulties or regional instability. Furthermore, changes in currency fluctuations, shipping costs, or import tariffs could adversely affect cost of goods sold. Any of the above factors could cause Savara to delay or suspend anticipated or ongoing trials, regulatory submissions or commercialization of its product candidates, entail higher costs or result in Savara being unable to effectively commercialize its products. Savara’s dependence upon third parties for the manufacture of its clinical trial
49
Table of Contents
material may adversely affect its future costs and its ability to develop and commercialize its product candidates on a timely and competitive basis.
Savara relies significantly on third parties to conduct its nonclinical testing and clinical studies and other aspects of its development programs and if those third parties do not satisfactorily perform their contractual obligations or meet anticipated deadlines, the development of its product candidates could be adversely affected.
Savara does not employ personnel or possess the facilities necessary to conduct many of the activities associated with its programs. Savara engages consultants, advisors, CROs, CMOs and others to assist in the design and conduct of nonclinical and clinical studies of its product candidates, with interpretation of the results of those studies and with regulatory activities, and Savara expects to continue to outsource all or a significant amount of such activities. As a result, many important aspects of its development programs are and will continue to be outside its direct control, and its third-party service providers may not perform their activities as required or expected including the maintenance of GCP, GLP and GMP compliance, which are ultimately Savara’s responsibility to ensure. Further, such third parties may not be as committed to the success of Savara’s programs as Savara’s own employees and, therefore, may not devote the same time, thoughtfulness or creativity to completing projects or problem-solving as Savara’s own employees would. To the extent Savara is unable to successfully manage the performance of third-party service providers, its business may be adversely affected.
The CROs that Savara engages to execute its clinical studies play a significant role in the conduct of the studies, including the collection and analysis of study data, and Savara likely will depend on CROs and clinical investigators to conduct future clinical studies and to assist in analyzing data from completed studies and developing regulatory strategies for its product candidates. Individuals working at the CROs with which it contracts, as well as investigators at the sites at which its studies are conducted, are not Savara’s employees, and Savara has limited control over the amount or timing of resources that they devote to their programs. If Savara’s CROs, study investigators, and/or third-party sponsors fail to devote sufficient time and resources to studies of its product candidates, if Savara and/or its CROs do not comply with all GLP and GCP regulatory and contractual requirements, or if their performance is substandard, it may delay commencement and/or completion of these studies, submission of applications for regulatory approval, regulatory approval, and commercialization of its product candidates. Failure of CROs to meet their obligations to Savara could adversely affect development of its product candidates.
In addition, CROs Savara engages may have relationships with other commercial entities, some of which may compete with Savara. Through intentional or unintentional means, Savara’s competitors may benefit from lessons learned on the Savara project that could ultimately harm Savara’s competitive position. Moreover, if a CRO fails to properly, or at all, perform its activities during a clinical study, Savara may not be able to enter into arrangements with alternative CROs on acceptable terms or in a timely manner, or at all. Switching CROs may increase costs and divert management time and attention. In addition, there likely would be a transition period before a new CRO commences work. These challenges could result in delays in the commencement or completion of Savara’s clinical studies, which could materially impact its ability to meet its desired and/or announced development timelines and have a material adverse impact on its business and financial condition.
Savara currently has limited marketing capabilities and no sales organization. If Savara is unable to establish sales and marketing capabilities on its own or through third parties, it will be unable to successfully commercialize its products, if approved, or generate product revenue.
To commercialize Savara’s products, if approved, in the United States and other jurisdictions it seeks to enter, Savara must build its marketing, sales, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and it may not be successful in doing so. If Savara’s products receive regulatory approval, it expects to market such products in the United States through a focused, specialized sales force, which will be costly and time consuming. Savara has no prior experience in the
50
Table of Contents
marketing and sale of pharmaceutical products and there are significant risks involved in building and managing a sales organization, including its ability to hire, retain and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel and effectively manage a geographically dispersed sales and marketing team. Outside of the United States, Savara may consider collaboration arrangements. If Savara is unable to enter into such arrangements on acceptable terms or at all, it may not be able to successfully commercialize its products in certain markets. Any failure or delay in the development of its internal sales, marketing and distribution capabilities would adversely impact the commercialization of its products. If Savara is not successful in commercializing its products, either on its own or through collaborations with one or more third parties, its future product revenue will suffer and it would incur significant additional losses.
Savara is the process of integrating the systems, people and contracts from the recent acquisition of Serendex and the complete scope and impact of the integration is unknown.
Savara’s acquisition of the assets of Serendex Pharmaceuticals A/S on July 15, 2016 has inherent risks, including risks associated with the integration of operations, systems and personnel. Savara has devoted its resources towards the successful integration of the companies, but there is potential exposure to unknown or contingent liabilities of the acquired company, the possible loss of key employees, liability associated with the assumption of legacy agreements, and many other such risks typical for such acquisitions.
To establish a sales and marketing infrastructure and expand its manufacturing capabilities, Savara will need to increase the size of its organization, and Savara may experience difficulties in managing this growth.
As of December 31, 2016, Savara had 15 full-time employees, including 10 employees engaged in research and development. As Savara advances its product candidates through the development process and to commercialization, it will need to continue to expand its development, regulatory, quality, managerial, sales and marketing, operational, finance and other resources to manage its operations and clinical trials, continue its development activities and commercialize its product candidates, if approved. As its operations expand, Savara expects that it will need to manage additional relationships with various manufacturers and collaborative partners, suppliers and other organizations.
Due to Savara’s limited financial resources and its limited experience in managing a company with such anticipated growth, Savara may not be able to effectively manage the expansion of its operations or recruit and train additional qualified personnel. In addition, the physical expansion of its operations may lead to significant costs and may divert its management and resources. Any inability to manage growth could delay the execution of its development and strategic objectives, or disrupt its operations, which could materially impact its business, revenue and operating results.
Savara’s product candidates may cause undesirable side effects or adverse events, or have other properties that could delay or prevent its clinical development, regulatory approval or commercialization.
Undesirable side effects or adverse events caused by Savara’s product candidates could interrupt, delay or halt clinical studies and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all indications, and in turn prevent Savara from commercializing its product candidates. A significant challenge in clinical development is that the patient population in early studies, where small numbers of patients are required, is different to the patient population observed in later stage studies, where larger groups of patients are required. For example, patients in earlier stage studies may be more sick, compliant, or otherwise motivated than patients in larger studies. As such, efficacy or safety results may differ significantly between studies. Side-effects seen at high doses in earlier studies of AeroVanc, such as bronchoconstriction or other airway irritation, may be seen in significant numbers at the lower doses selected for later studies. Also, for AeroVanc, while not observed in the Phase 2 clinical study, the emergence of vancomycin-resistant MRSA could occur during the longer dosing period of AeroVanc that is currently planned for the Phase 3 clinical study. If this
51
Table of Contents
or other undesirable side effects occur, they could possibly prevent approval, which would have a material and adverse effect on its business.
If any of its product candidates receive marketing approval and Savara or others later identify undesirable side effects caused by the product:
| • | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; |
| • | regulatory authorities may withdraw its approval of the product; |
| • | Savara may be required to change the way the product is administered, conduct additional clinical studies or change the labeling of the product; and |
| • | its reputation may suffer. |
Any of these events could prevent Savara from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product, which in turn could delay or prevent Savara from generating significant revenue from its sale.
Savara may not achieve its projected development goals in the time frames Savara has announced.
Savara has set goals for accomplishing certain objectives material to the successful development of its product candidates. The actual timing of these events may vary due to many factors, including delays or failures in its nonclinical testing, clinical studies and manufacturing and regulatory activities and the uncertainties inherent in the regulatory approval process. From time to time Savara creates estimates for the completion of enrollment of or announcement of data from clinical studies of its product candidates. However, predicting the rate of enrollment or the time from completion of enrollment to announcement of data for any clinical study requires Savara to make significant assumptions that may prove to be incorrect. As discussed in other risk factors above, its estimated enrollment rates and the actual rates may differ materially and the time required to complete enrollment of any clinical study may be considerably longer than Savara estimates. Such delays may adversely affect its financial condition and results of operations.
Even if Savara completes a clinical study with successful results, Savara may not achieve its projected development goals in the time frames Savara initially anticipates or announces. If a development plan for a product candidate becomes more extensive and costly than anticipated, Savara may determine that the associated time and cost are not financially justifiable and, as a result, may discontinue development in a particular indication or of the product candidate as a whole. In addition, even if a study did complete with successful results, changes may occur in regulatory requirements or policy during the period of product development and/or regulatory review of an NDA that relate to the data required to be included in NDAs which may require additional studies that may be costly and time consuming. Any of these actions may be viewed negatively, which could adversely impact its financial condition.
Further, throughout development, Savara must provide adequate assurance to the FDA and other regulatory authorities that Savara can consistently develop and produce its product candidates in conformance with GLP, GCP, cGMP, and other regulatory standards. As discussed above, Savara relies on CMOs for the manufacture of clinical, and future commercial, quantities of its product candidates. If future FDA or other regulatory authority inspections identify cGMP compliance deficiencies at these third-party facilities, production of its clinical trial material or, in the future, commercial product, could be disrupted, causing potentially substantial delay in or failure of development or commercialization of its product candidates.
52
Table of Contents
Savara’s employees, independent contractors and consultants, principal investigators, CROs, CMOs and other vendors, and any future commercial partners may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements, which could cause significant liability for Savara and harm its reputation.
Savara is exposed to the risk that its employees, independent contractors and consultants, principal investigators, CROs, CMOs and other vendors, and any future commercial partners may engage in fraudulent conduct or other misconduct, including intentional failures to comply with FDA regulations or similar regulations of comparable foreign regulatory authorities, to provide accurate information to the FDA or comparable foreign regulatory authorities, to comply with manufacturing standards required by cGMP or Savara standards, to comply with federal and state healthcare fraud and abuse laws and regulations and similar laws and regulations established and enforced by comparable foreign regulatory authorities, and to report financial information or data accurately or disclose unauthorized activities to them. The misconduct of its employees and other Savara service providers could involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and serious harm to its reputation. Savara intends to adopt a code of business ethics and conduct, but it is not always possible to identify and deter such misconduct, and the precautions Savara takes to detect and prevent this activity, such as the implementation of a quality system which entails vendor audits by quality experts, may not be effective in controlling unknown or unmanaged risks or losses or in protecting Savara from governmental investigations or other actions or lawsuits stemming from a failure to be in compliance with such laws or regulations. If any such actions are instituted against them, and Savara is not successful in defending itself or asserting its rights, those actions could have a significant impact on its business and results of operations, including the imposition of significant fines or other sanctions. For example, if one of its manufacturing partners was placed under a consent decree, Savara may be hampered in its ability to manufacture clinical or commercial supplies.
Savara’s business and operations would suffer in the event of third-party computer system failures, cyber-attacks on third-party systems or deficiency in its cyber security.
Savara relies on information technology systems, including third-party “cloud based” service providers, to keep financial records, maintain laboratory, clinical data and corporate records, communicate with staff and external parties and operate other critical functions. This includes critical systems such as email, other communication tools, electronic document repositories, and archives. If any of these third-party information technology (IT) providers are compromised due to computer viruses, unauthorized access, malware, natural disasters, fire, terrorism, war and telecommunication failures, electrical failures, cyber-attacks or cyber-intrusions over the internet, then sensitive emails or documents could be exposed or deleted. Similarly, Savara could incur business disruption if its access to the internet is compromised and Savara is unable to connect with third-party IT providers. The risk of a security breach or disruption, particularly through cyber-attacks or cyber intrusion, including by computer hackers, foreign governments, and cyber terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. In addition, Savara relies on those third parties to safeguard important confidential personal data regarding its employees and patients enrolled in its clinical trials. If a disruption event were to occur and cause interruptions in a third-party IT provider’s operations, it could result in a disruption of its drug development programs. For example, the loss of clinical trial data from completed, ongoing or planned clinical trials could result in delays in its regulatory approval efforts and significantly increase its costs to recover or reproduce the data. To the extent that any disruption or security breach results in a loss of or damage to its data or applications, or inappropriate disclosure of confidential or proprietary information, Savara could incur liability and development of its product candidates could be delayed, or could fail.
Savara’s operations might be interrupted by the occurrence of a natural disaster or other catastrophic event.
Savara’s corporate headquarters is located in a single commercial facility in Austin, Texas, USA. Savara maintains a second office in a single commercial facility in Denmark where many of Savara’s product
53
Table of Contents
development staff are located. Important documents and records, including copies of its regulatory documents and other records for its product candidates, are located both at a secure offsite document storage facility as well at its own facilities and Savara depends on its facilities for the continued operation of its business. Natural disasters and other catastrophic events, such as wildfires and other fires, earthquakes and extended power interruptions and terrorist attacks or severe weather conditions, could significantly disrupt its operations and result in additional, unplanned expense. As a small company with limited resources, Savara has not prepared or implemented a formal business continuity or disaster recovery plan and any natural disaster or catastrophic event could disrupt its business operations and result in setbacks to its development programs. Even though Savara believes it carries commercially reasonable insurance, Savara might suffer losses that are not covered by or exceed the coverage available under these insurance policies.
Risks Related to Drug Development and Commercialization
Savara depends on the successful completion of clinical studies of its product candidates, and any positive results in prior clinical studies do not ensure that ongoing or future clinical studies will be successful.
Pharmaceutical products are subject to stringent regulatory requirements covering quality, safety, and efficacy. The burden of proof is on the manufacturer, such as Savara, to show with substantial clinical data that the risk/benefit profile for any new drug is favorable. Only after successfully completing extensive pharmaceutical development, nonclinical testing, and clinical studies may a product be considered for regulatory approval.
Clinical studies are expensive, difficult to design and implement, they can take many years to complete, and outcomes are inherently uncertain. A drug product may fail to demonstrate positive results at any stage of testing despite having progressed satisfactorily through nonclinical testing and initial clinical studies. There is significant risk in clinical development where later stage clinical studies are designed and powered based on the analysis of data from earlier studies, with these earlier studies involving a smaller number of patients, and the results of the earlier studies being driven primarily by a subset of responsive patients. In addition, interim results of a clinical study do not necessarily predict final results. Further, clinical study data frequently are susceptible to varying interpretations. Medical professionals and/or regulatory authorities may analyze or weigh study data differently than the sponsor company, resulting in delay or failure to obtain marketing approval for a product candidate. Additionally, the possible lack of standardization across multiple investigative sites may induce variability in the results which can interfere with the evaluation of treatment effects.
If Savara licenses rights to develop its product candidates to independent third parties or otherwise permit such third parties to evaluate its product candidates in clinical studies, Savara may have limited control over those clinical studies. Any safety or efficacy concern identified in a third-party sponsored study could adversely affect its or another licensee’s development of its product candidate and prospects for its regulatory approval, even if the data from that study are subject to varying interpretations and analyses.
There is significant risk that ongoing and future clinical studies of its product candidates are unsuccessful. Negative or inconclusive results could cause the FDA and other regulatory authorities to require Savara to repeat or conduct additional clinical studies, which could significantly increase the time and expense associated with development of that product candidate or cause Savara to elect to discontinue one or more clinical programs. Failure to complete a clinical study of a product candidate or an unsuccessful result of a clinical study could have a material adverse effect on its business.
Both of Savara’s product candidates have received Orphan Drug Designation by the Food and Drug Administration (FDA) and Molgradex has received Orphan Drug Designation also in Europe. While orphan designation provides certain benefits there are also associated risks.
AeroVanc has been granted Orphan Drug Designation in the United States by the FDA for the treatment of persistent methicillin-resistant Staphylococcus aureus (MRSA) lung infection in patients with cystic fibrosis and
54
Table of Contents
Molgradex has received Orphan Drug Designation in the United States by the FDA and in Europe by the European Medicines Agency for the treatment of pulmonary alveolar proteinosis (PAP). Orphan Designation will not shorten the regulatory review or reduce the clinical data requirements needed to obtain approval. If approval is received to market either AeroVanc or Molgradex for the respective indications, FDA will not approve a similar product, with the same active ingredient, to AeroVanc or Molgradex for seven years and the European Medicines Agency will not approve a similar product to Molgradex for ten years, unless Savara is unable to produce enough supply to meet demand in the marketplace or another similar product, with the same active ingredient, is deemed clinically superior. Similar product candidates, with the same active ingredient and route of delivery, may be granted Orphan Drug Designation during the development of the respective products, but the Orphan Drug exclusivity is granted only to the first of such products approved, which means there is risk that a competitor product candidate may receive approval and Orphan Drug exclusivity before Savara, thus preventing Savara from marketing one or more of its product candidates until the exclusivity of the competing product expires. Also, the Orphan Drug status will not prevent a competitor with a different active ingredient from competing with Savara’s product candidates. If Savara is prevented from marketing one or more product candidates due to a competitor’s Orphan Drug exclusivity, this would have a material adverse effect on its business.
Delays in commencement and completion of clinical studies are common and have many causes. Delays in clinical studies of Savara’s product candidates could increase overall development costs and jeopardize its ability to obtain regulatory approval and successfully commercialize any approved products.
Clinical testing typically is expensive, can take many years to complete, and its outcome is inherently uncertain. Clinical studies may not commence on time or be completed on schedule, if at all. The commencement and completion of clinical studies can be delayed for a variety of reasons, including:
| • | inability to raise sufficient funding to initiate or continue a clinical study; |
| • | delays in obtaining regulatory approval to commence a clinical study; |
| • | delays in identifying and reaching agreement on acceptable terms with prospective contract research organizations, or CROs, and clinical study sites and investigators, which agreements can be subject to extensive negotiation and may vary significantly among study sites; |
| • | delays in obtaining regulatory approval in a prospective country; |
| • | delays in obtaining ethic committee approval to conduct a clinical study at a prospective site; |
| • | delays in reaching agreements on acceptable terms with prospective contract manufacturing organizations, or CMOs, or other vendors for the production and supply of clinical trial material and, if necessary, drug administration devices, which agreements can be subject to extensive negotiation; |
| • | delays in the production or delivery of sufficient quantities of clinical trial material or drug delivery devices from its CMOs and other vendors to initiate or continue a clinical study; |
| • | delays due to product candidate recalls as result of stability failure, excessive product complaints or other failures of the product candidate during its use or testing; |
| • | invalidation of clinical data caused by premature unblinding or integrity issues; |
| • | invalidation of clinical data caused by mixing up of the active drug and placebo through randomization or manufacturing errors; |
| • | delays on the part of its CROs, CMOs, and other third-party contractors in developing procedures and protocols or otherwise conducting activities in accordance with applicable policies and procedures and in accordance with agreed upon timelines; |
| • | delays in identifying and hiring or engaging, as applicable, additional employees or consultants to assist in managing clinical study-related activities; |
55
Table of Contents
| • | delays in recruiting and enrolling individuals to participate in a clinical study, which historically can be challenging in orphan diseases; |
| • | delays caused by patients dropping out of a clinical study due to side effects, concurrent disorders, difficulties in adhering to the study protocol, unknown issues related to different patient profiles than in previous studies, such as the reduced age limit required for inclusion into the planned AeroVanc Phase 3 study, or otherwise; |
| • | delays in having patients complete participation in a clinical study, including returning for post-treatment follow-up; |
| • | delays resulting from study sites dropping out of a trial, providing inadequate staff support for the study, problems with shipment of study supplies to clinical sites or focusing its staff’s efforts on enrolling studies that compete for the same patient population; |
| • | suspension of enrollment at a study site or the imposition of a clinical hold by the FDA or other regulatory authority following an inspection of clinical study operations at study sites or finding of a drug-related serious adverse event; and |
| • | delays in quality control/quality assurance procedures necessary for study database lock and analysis of unblinded data. |
Patient enrollment, a critical component to successful completion of a clinical study, is affected by many factors, including the size and nature of the study population, the proximity of patients to clinical sites, the eligibility criteria for the study, the design of the clinical study, ongoing studies competing for the same patient population and clinicians’, patients’ perceptions as to the potential advantages of the drug being studied in relation to available alternatives, including therapies being investigated by other companies which may be viewed as more beneficial or important to study, fear of being randomized to the placebo arm, and changes in standard of care. Challenges to complete enrollment can be exacerbated in orphan indications, like those being pursued by Savara, with a limited number of qualifying patients and the lack of clinical sites with the necessary expertise and experience to conduct Savara’s studies. Further, completion of a clinical study and/or its results may be adversely affected by failure to retain patients who enroll in a study but withdraw due to adverse side effects, perceived lack of efficacy, belief that they are on placebo, improvement in condition before treatment has been completed, or for personal reasons, or without reason, or by patients who fail to return for or complete post-treatment follow-up.
Clinical studies may not begin on time or be completed in the time frames Savara anticipates and may be costlier than Savara anticipates for a variety of reasons, including one or more of those described above. The length of time necessary to successfully complete clinical studies varies significantly and is difficult to predict accurately. Savara may make statements regarding anticipated timing for completion of enrollment in and/or availability of results from its clinical studies, but such predictions are subject to a number of significant assumptions and actual timing may differ materially for a variety of reasons, including patient enrollment rates, length of time needed to prepare raw study data for analysis and then to review and analyze it, and other factors described above. If Savara experiences delays in the completion of a clinical study, if a clinical study is terminated, or if failure to conduct a study in accordance with regulatory requirements or the study’s protocol leads to deficient safety and/or efficacy data, the regulatory approval and/or commercial prospects for its product candidates may be harmed and its ability to generate product revenue will be delayed. In addition, any delays in completing its clinical studies likely will increase its development costs. Further, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical studies may ultimately lead to the denial of regulatory approval of a product candidate. Even if Savara ultimately commercializes its product candidates, the standard of care may have changed or other therapies for the same indications may have been introduced to the market in the interim and may establish a competitive threat to Savara or diminish the need for Savara’s products.
56
Table of Contents
Clinical studies are very expensive, difficult to design and implement, often take many years to complete, and the outcome is inherently uncertain.
Clinical development of pharmaceutical products for humans is generally very expensive, takes many years to complete and failures can occur at any stage of clinical testing. Savara estimates that clinical development of its product candidates will take several additional years to complete, but because of the variety of factors that can affect the design, timing and outcome of clinical studies, Savara is unable to estimate the exact funds required to complete research and development, obtain regulatory approval and commercialize all of its product candidates. Savara will need significant additional capital to continue to advance its products as per current business plans.
Failure at any stage of clinical testing is not uncommon and Savara may encounter problems that would require additional, unplanned studies or cause Savara to abandon a clinical development program.
In addition, a clinical study may be suspended or terminated by Savara, an IRB, a data safety monitoring board, the FDA or other regulatory authorities due to a number of factors, including:
| • | lack of adequate funding to continue the study; |
| • | failure to conduct the study in accordance with regulatory requirements or the study’s protocol; |
| • | inspection of clinical study operations or sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| • | unforeseen safety issues, including adverse side effects; or |
| • | changes in governmental regulations or administrative actions. |
Changes in governmental regulations and guidance relating to clinical studies may occur and Savara may need to amend study protocols to reflect these changes, or Savara may amend study protocols for other reasons. Amendments may require Savara to resubmit protocols to IRBs for reexamination and approval or renegotiate terms with CROs, study sites and investigators, all of which may adversely impact the costs or timing of or its ability to successfully complete a trial.
There is significant uncertainty regarding the regulatory approval process for any investigational new drug, substantial further testing and validation of its product candidates and related manufacturing processes may be required, and regulatory approval may be conditioned, delayed or denied, any of which could delay or prevent Savara from successfully marketing its product candidates and substantially harm its business.
Pharmaceutical products generally are subject to rigorous nonclinical testing and clinical studies and other approval procedures mandated by the FDA and foreign regulatory authorities. Various federal and foreign statutes and regulations also govern or materially influence the manufacturing, safety, labeling, storage, record keeping and marketing of pharmaceutical products. The process of obtaining these approvals and the subsequent compliance with appropriate U.S. and foreign statutes and regulations is time-consuming and requires the expenditure of substantial resources.
Savara is preparing AeroVanc for a Phase 3 trial, the success of which will be needed for FDA approval to market AeroVanc in the United States to treat persistent MRSA lung infection in cystic fibrosis patients. While significant communication with the FDA on the Phase 3 study design has occurred, even if the Phase 3 clinical study meets all of its statistical goals and protocol end points, the FDA may not view the results as robust and convincing. They may require additional clinical studies and/or other costly studies, which could require Savara to expend substantial additional resources and could significantly extend the timeline for clinical development prior to market approval. Additionally, Savara is required by the FDA to conduct a two-year nonclinical carcinogenicity study on the AeroVanc powder. The results of this study will not be known until a short time prior to potential submission of an NDA for AeroVanc. If the carcinogenicity study cannot be completed for technical or other reasons, or provides results that the FDA determine to be concerning, this may cause a delay or failure in obtaining approval for AeroVanc.
57
Table of Contents
Molgradex is currently undergoing a Phase 2/3 clinical study in Europe and Japan. Concurrently, Savara plans to make formulation changes to Molgradex that would simplify the composition of the drug product and eliminate potentially harmful excipients. While this change is expected by Savara to reduce studies and/or other documentation requirements, the regulatory agencies may require additional clinical or nonclinical studies prior to approval, even if current clinical studies are deemed successful, which could require Savara to expend substantial additional resources and significantly extend the timeline for clinical development of Molgradex in PAP.
Savara is currently undergoing active discussion with the FDA on the requirements for obtaining IND approval to initiate clinical studies in the United States and achieve NDA approval for Molgradex. However, no agreement has yet been reached on the design of the clinical program required for the submission of an NDA, and there is risk that reaching agreement may take longer than currently planned, or the FDA may require such studies that Savara deems unfeasible, preventing Savara to reach agreement with the FDA, which may result in delay or failure to complete the development of Molgradex in the US.
Significant uncertainty exists with respect to the regulatory approval process for any investigational new drug, including AeroVanc and Molgradex. Regardless of any guidance the FDA or foreign regulatory agencies may provide a drug’s sponsor during its development, the FDA or foreign regulatory agencies retains complete discretion in deciding whether to accept an NDA or the equivalent foreign regulatory approval submission for filing or, if accepted, approve an NDA. There are many components to an NDA or marketing authorization application submission in addition to clinical study data. For example, the FDA or foreign regulatory agencies will review the sponsor’s internal systems and processes, as well as those of its CROs, CMOs and other vendors, related to development of its product candidates, including those pertaining to its clinical studies and manufacturing processes. Before accepting an NDA for review or before approving the NDA, the FDA or foreign regulatory agencies may request that Savara provide additional information that may require significant resources and time to generate and there is no guarantee that its product candidates will be approved for any indication for which Savara may apply. The FDA or foreign regulatory agencies may choose not to approve an NDA for any of a variety of reasons, including a decision related to the safety or efficacy data, manufacturing controls or systems, or for any other issues that the agency may identify related to the development of its product candidates. Even if one or more Phase 3 clinical studies are successful in providing statistically significant evidence of the efficacy and safety of the investigational drug, the FDA or foreign regulatory agencies may not consider efficacy and safety data from the submitted studies adequate scientific support for a conclusion of effectiveness and/or safety and may require one or more additional Phase 3 or other studies prior to granting marketing approval. If this were to occur, the overall development cost for the product candidate would be substantially greater and its competitors may bring products to market before Savara, which could impair its ability to generate revenues from the product candidates, or even seek approval, if blocked by a competitor’s Orphan Drug exclusivity, which would have a material adverse effect on Savara’s business, financial condition and results of operations.
Further, development of Savara’s product candidates and/or regulatory approval may be delayed for reasons beyond its control. For example, U.S. federal government shut-down or budget sequestration, such as one that occurred during 2013, may result in significant reductions to the FDA’s budget, employees and operations, which may lead to slower response times and longer review periods, potentially affecting Savara’s ability to progress development of its product candidates or obtain regulatory approval for its product candidates.
Even if the FDA or foreign regulatory agencies grant approvals for Savara’s product candidates, the conditions or scope of the approval(s) may limit successful commercialization of the product candidates and impair Savara’s ability to generate substantial sales revenue. For example, the FDA may approve label claims for AeroVanc with age restrictions and/or treatment duration limitations, or Molgradex with restrictions for use only by patients unresponsive to the current standard of care. They may limit the label of AeroVanc or Molgradex to a subset of patients based on a review of which patient groups had the greatest efficacious response in clinical studies. Such label restriction may be undesirable and may limit successful commercialization. The FDA or foreign regulatory agencies may also only grant marketing approval contingent on the performance of costly
58
Table of Contents
post-approval nonclinical or clinical studies, or subject to warnings or contraindications that limit commercialization. Additionally, even after granting approval, the manufacturing processes, labeling, packaging, distribution, adverse event reporting, storage, advertising, promotion and recordkeeping for its products will be subject to extensive and ongoing regulatory requirements. These requirements include submissions of safety and other post-marketing information and reports, registration, and continued compliance with current good manufacturing processes, or cGMP, good clinical practices, international conference on harmonization regulations and good laboratory practices, which are regulations and guidelines that are enforced by the FDA or foreign regulatory agencies for all of its clinical development and for any clinical studies that Savara conducts post-approval. The FDA or foreign regulatory agencies may decide to withdraw approval, add warnings or narrow the approved indications in the product label, or establish risk management programs that could restrict distribution of its products. These actions could result from, among other things, safety concerns, including unexpected side effects or drug-drug interaction problems, or concerns over misuse of a product. If any of these actions were to occur following approval, Savara may have to discontinue commercialization of the product, limit its sales and marketing efforts, implement risk minimization procedures, and/or conduct post-approval studies, which in turn could result in significant expense and delay or limit its ability to generate sales revenues.
Regulations may be changed prior to submission of an NDA that require higher hurdles than currently anticipated. These may occur as a result of drug scandals, recalls, or a political environment unrelated to Savara’s products.
Even if Savara receives regulatory approval for a product candidate, Savara may face regulatory difficulties that could materially and adversely affect its business, financial condition and results of operations.
Even if initial regulatory approval is obtained, as a condition to the initial approval the FDA or a foreign regulatory agency may impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies or marketing surveillance programs, any of which would limit the commercial potential of the product. Its product candidates also will be subject to ongoing FDA requirements related to the manufacturing processes, labeling, packaging, storage, distribution, advertising, promotion, record-keeping and submission of safety and other post-market information regarding the product. For instance, the FDA may require changes to approved drug labels, require post-approval clinical studies and impose distribution and use restrictions on certain drug products. In addition, approved products, manufacturers and manufacturers’ facilities are subject to continuing regulatory review and periodic inspections. If previously unknown problems with a product are discovered, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, the FDA may impose restrictions on that product or Savara, including requiring withdrawal of the product from the market. If Savara or a CMO of Savara’s fail to comply with applicable regulatory requirements, a regulatory agency may:
| • | issue warning letters or untitled letters; |
| • | impose civil or criminal penalties; |
| • | suspend or withdraw regulatory approval; |
| • | suspend or terminate any ongoing clinical studies; |
| • | refuse to approve pending applications or supplements to approved applications; |
| • | exclude its product from reimbursement under government healthcare programs, including Medicaid or Medicare; |
| • | impose restrictions or affirmative obligations on Savara’s or its CMO’s operations, including costly new manufacturing requirements; |
| • | close the facilities of a CMO; or |
| • | seize or detain products or require a product recall. |
59
Table of Contents
If any of Savara’s product candidates for which Savara receive regulatory approval fails to achieve significant market acceptance among the medical community, patients or third-party payers, the revenue Savara generates from its sales will be limited and its business may not be profitable.
Savara’s success will depend in substantial part on the extent to which its product candidates, if approved, are accepted by the medical community and patients and reimbursed by third-party payers, including government payers. The degree of market acceptance with respect to each of its approved products, if any, will depend upon a number of factors, including:
| • | the safety and efficacy of its products as demonstrated in clinical studies; |
| • | acceptance in the medical and patient communities of its products as a safe and effective treatment; |
| • | the product’s taste, ease of use, or features associated with the delivery device; |
| • | the perceived advantages of its product over alternative treatments, including with respect to the incidence and severity of any adverse side effects and the cost of treatment; |
| • | the indications for which its product is approved; |
| • | claims or other information (including limitations or warnings) in its product’s approved labeling; |
| • | reimbursement and coverage policies of government and other third-party payers; |
| • | pricing and cost-effectiveness of its product relative to alternative treatments; |
| • | availability of alternative treatments; |
| • | smaller than expected market size due to lack of disease awareness of a rare disease, or the patient population with a specific rare disease being smaller than anticipated; |
| • | inappropriate diagnostic efforts due to limited knowledge and/or resources among clinicians; |
| • | the prevalence of off-label substitution of chemically equivalent products or alternative treatments; and |
| • | the resources Savara devotes to marketing its product and restrictions on promotional claims Savara can make with respect to the product. |
Savara cannot predict with reasonable accuracy whether physicians, patients, healthcare insurers or health maintenance organizations, or the medical community in general, will accept or utilize any of its products, if approved. If its product candidates are approved but do not achieve an adequate level of acceptance by these parties, Savara may not generate sufficient revenue to become or remain profitable. In addition, its efforts to educate the medical community and third-party payers regarding benefits of its products may require significant resources and may never be successful.
If Savara determines that a product candidate may not achieve adequate market acceptance or that the potential market size does not justify additional expenditure on the program, Savara may reduce its expenditures on the development and/or the process of seeking regulatory approval of the product candidate while Savara evaluates whether and on what timeline to move the program forward.
Even if Savara receives regulatory approval to market one or more of its product candidates in the United States, Savara may never receive approval or commercialize its products outside of the United States, which would limit its ability to realize the full commercial potential of its product candidates.
In order to market products outside of the United States, Savara must establish and comply with the numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and validation and additional administrative review periods. The time required to obtain approval in other countries generally differs from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks
60
Table of Contents
detailed above regarding FDA approval in the United States, as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects detailed above regarding FDA approval in the United States. As described above, such effects include the risks that its product candidates may not be approved for all indications requested, which could limit the uses of its product candidates and have an adverse effect on product sales, and that such approval may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
Conversely, if the product candidates do receive approval outside the US in the future, Savara may not meet the FDA requirements in the United States for approval. For example, Molgradex is currently being studied in Europe and Japan in what could be a pivotal study for use of Molgradex to treat PAP. However, in the United States, Savara does not yet have approval from the FDA to start clinical studies with Molgradex due to different requirements by the FDA, which have not yet been met or agreed upon.
Savara must comply with the U.S. Foreign Corrupt Practices Act and similar foreign anti-corruption laws.
The U.S. Foreign Corrupt Practices Act, to which Savara is subject, prohibits corporations and individuals from engaging in certain activities to obtain or retain business or to influence a person working in an official capacity. It is illegal to pay, offer to pay or authorize the payment of anything of value to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business or to otherwise influence a person working in an official capacity. Other countries, such as the U.K., have similar laws with which Savara must comply. Savara faces the risk that an employee or agent could be accused of violating one or more of these laws, particularly in geographies where significant overlap exists between local government and healthcare industries. Such an accusation, even if unwarranted, could prove disruptive to Savara’s developmental and commercialization efforts.
Risks Related to Savara’s Intellectual Property
Savara’s success will depend in part on obtaining and maintaining effective patent and other intellectual property protection for its product candidates and proprietary technology.
AeroVanc has received a U.S. Patent Notice of Allowance for its formulation in the United States, AeroVanc’s primary market. AeroVanc has either been issued patents or is prosecuting patent applications in numerous countries outside the United States. Savara has no patent protection for Molgradex for the treatment of PAP, and primarily relies on the Orphan Drug exclusivity as its primary barrier to competition. Both AeroVanc and Molgradex utilize proprietary delivery devices with exclusive supply agreements. Molgradex is eligible for protection via a proprietary cell bank used in the production of the drug substance. However, Savara’s success will depend in part on its ability to:
| • | obtain and maintain patent and other exclusivity with respect to Savara’s products and its uses; |
| • | prevent third parties from infringing upon its proprietary rights; |
| • | maintain proprietary know-how and trade secrets; |
| • | operate without infringing upon the patents and proprietary rights of others; and |
| • | obtain appropriate licenses to patents or proprietary rights held by third parties if infringement would otherwise occur or if necessary to secure exclusive rights to them, both in the United States and in foreign countries. |
The patent and intellectual property positions of biopharmaceutical companies generally are highly uncertain, involve complex legal and factual questions, and have been and continue to be the subject of much litigation. There is no guarantee that Savara has or will develop or obtain the rights to products or processes that
61
Table of Contents
are patentable, that patents will issue from any pending applications or that claims allowed will be sufficient to protect the technology Savara develops or have developed or that is used by Savara, its CMOs or its other service providers. In addition, any patents that are issued to Savara may be limited in scope or challenged, invalidated, infringed or circumvented, including by its competitors, and rights Savara have under issued patents may not provide competitive advantages to Savara. If competitors can develop and commercialize technology and products similar to Savara’s, its ability to successfully commercialize its technology and products may be impaired.
Patent applications in the United States are confidential for a period of time until they are published, and publication of discoveries in scientific or patent literature typically lags actual discoveries by several months. As a result, Savara cannot be certain that the inventors listed in any patent or patent application owned by Savara were the first to conceive of the inventions covered by such patents and patent applications (for U.S. patent applications filed before March 16, 2013), or that such inventors were the first to file patent applications for such inventions outside the United States and, after March 15, 2013, in the United States. In addition, changes in or different interpretations of patent laws in the United States and foreign countries may affect Savara’s patent rights and limit the number of patents Savara can obtain, which could permit others to use its discoveries or to develop and commercialize Savara’s technology and products without any compensation to Savara.
Savara’s AeroVanc patent is specific to the formulation of the AeroVanc powder. While this may prevent identical products from entering the market, it may not preclude someone skilled in the art from inventing an alternate formulation approach with comparable or improved characteristics.
Savara also relies on unpatented know-how and trade secrets and continuing technological innovation to develop and maintain its competitive position, which Savara seeks to protect, in part, through confidentiality agreements with employees, consultants, collaborators and others. Savara also has invention or patent assignment agreements with its employees and certain consultants. The steps Savara have taken to protect its proprietary rights, however, may not be adequate to preclude misappropriation of or otherwise protect its proprietary information or prevent infringement of its intellectual property rights, and Savara may not have adequate remedies for any such misappropriation or infringement. In addition, it is possible that inventions relevant to Savara’s business could be developed by a person not bound by an invention assignment agreement with Savara or independently discovered by a competitor.
Savara also intends to rely on regulatory exclusivity for protection of its product candidates, if approved for commercial sale. Implementation and enforcement of regulatory exclusivity, which may consist of regulatory data protection and market protection, varies widely from country to country. Failure to qualify for regulatory exclusivity, or failure to obtain or maintain the extent or duration of such protections that Savara expects for its product candidates, if approved, could affect its decision on whether to market the products in a particular country or countries or could otherwise have an adverse impact on its revenue or results of operations. For Molgradex, which is administered via nebulization, Savara may rely on regulatory exclusivity for the combination of Molgradex and its delivery system. However, there is no assurance that its Molgradex product and its delivery system, if approved, will benefit from this type of market protection.
Savara may rely on trademarks, trade names and brand names to distinguish its products, if approved for commercial sale, from the products of its competitors. Savara intends to seek approval for new names for AeroVanc and Molgradex that meet the FDA’s and foreign regulatory requirements. However, Savara’s trademark applications may not be approved. Third parties may also oppose Savara’s trademark applications or otherwise challenge its use of the trademarks in which case Savara may expend substantial resources to defend its proposed or approved trademarks and may enter into agreements with third parties that may limit Savara’s use of its trademarks. In the event that Savara’s trademarks are successfully challenged, Savara could be forced to rebrand its product, which could result in loss of brand recognition and could require Savara to devote significant resources to advertising and marketing these new brands. For example, Savara filed a trademark for the name “Savara” and was challenged. Savara decided to terminate its application, which it may revisit such filings at a
62
Table of Contents
future date. Further, Savara’s competitors may infringe its trademarks or Savara may not have adequate resources to enforce its trademarks.
Savara’s success depends on its ability to prevent competitors from duplicating or developing and commercializing equivalent versions of its product candidates, but patent protection may be difficult to obtain and any issued claims may be limited.
Savara has filed for patent protection in the United States and other countries to cover the formulation of AeroVanc and was granted a notice of allowance in the United States, its primary market. However, this patent may not provide Savara with significant competitive advantages, because the validity or enforceability of the patents may be challenged and, if instituted, one or more of the challenges may be successful. Patents may be challenged in the United States under post-grant review proceedings, inter partes reexamination, ex parte re-examination, or challenges in district court. Any patents issued in foreign jurisdictions may be subjected to comparable proceedings lodged in various foreign patent offices, or courts. These proceedings could result in either loss of the patent or loss or reduction in the scope of one or more of the claims of the patent. Even if a patent issues, and is held valid and enforceable, competitors may be able to design around Savara’s patents, such as by using pre-existing or newly developed technology, in which case competitors may not infringe Savara’s issued claims and may be able to market and sell products that compete directly with Savara’s before and after its patents expire.
The patent prosecution process is expensive and time-consuming. Savara and any future licensors and licensees may not apply for or prosecute patents on certain aspects of its product candidates at a reasonable cost, in a timely fashion, or at all. Savara may not have the right to control the preparation, filing and prosecution of some patent applications related to its product candidates or technologies. As a result, these patents and patent applications may not be prosecuted and enforced in a manner consistent with the best interests of Savara. It is also possible that Savara or any future licensors or licensees will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. Further, it is possible that defects of form in the preparation or filing of Savara’s patent applications may exist, or may arise in the future, such as with respect to proper priority claims, inventorship, assignment, or claim scope. If there are material defects in the form or preparation of its patents or patent applications, such patents or applications may be invalid or unenforceable. In addition, one or more parties may independently develop similar technologies or methods, duplicate its technologies or methods, or design around the patented aspects of its products, technologies or methods. Any of these circumstances could impair Savara’s ability to protect its products, if approved, in ways which may have an adverse impact on Savara’s business, financial condition and operating results.
Furthermore, the issuance of a patent is not conclusive as to its inventorship, scope, validity or enforceability, and Savara’s owned and licensed patents may be challenged in the courts or patent offices in and outside of the United States. Such challenges may result in loss of exclusivity or freedom to operate or in patent claims being narrowed, invalidated or held unenforceable, in whole or in part, which could limit Savara’s ability use its patents to stop others from using or commercializing similar or identical products or technology, or limit the duration of the patent protection of its technology and drugs. Given the amount of time required for the development, testing and regulatory review of new drug candidates, patents protecting such candidates might expire before or shortly after such candidates are commercialized. As a result, Savara’s owned and licensed patent portfolio may not provide Savara with sufficient rights to exclude others from commercializing drugs similar or identical to those of Savara once Orphan Drug and Qualified Infectious Disease Product exclusivities have expired. See the section entitled “Risks Related to Savara’s Industry” for further description of Orphan Drug and Qualified Infectious Disease Product exclusivities.
Enforcement of intellectual property rights in certain countries outside the United States, including China in particular, has been limited or non-existent. Future enforcement of patents and proprietary rights in many other countries will likely be problematic or unpredictable. Moreover, the issuance of a patent in one country does not
63
Table of Contents
assure the issuance of a similar patent in another country. Claim interpretation and infringement laws vary by nation, so the extent of any patent protection is uncertain and may vary in different jurisdictions.
Obtaining and maintaining patent protection depends on compliance with various procedural, document submission, fee payment and other requirements imposed by governmental patent agencies, and Savara’s patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance fees, renewal fees, annuity fees and various other governmental fees on patents and applications are required to be paid to the United States Patent and Trademark Office, or USPTO, and various governmental patent agencies outside of the United States in several stages over the lifetime of the patents and applications. The USPTO and various non-U.S. governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process and after a patent has issued. There are situations in which non-compliance can result in decreased patent term adjustment or in abandonment or lapse of the patent or patent application, leading to partial or complete loss of patent rights in the relevant jurisdiction.
Third parties may claim that Savara’s products, if approved, infringe on their proprietary rights and may challenge the approved use or uses of a product or its patent rights through litigation or administrative proceedings, and defending such actions may be costly and time consuming, divert management attention away from Savara’s business, and result in an unfavorable outcome that could have an adverse effect on Savara’s business.
Savara’s commercial success depends on its ability and the ability of its CMOs and component suppliers to develop, manufacture, market and sell its products and product candidates and use its proprietary technologies without infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which Savara is or may be developing products. Because patent applications can take many years to publish and issue, there currently may be pending applications, unknown to Savara, that may later result in issued patents that its products, product candidates or technologies infringe, or that the process of manufacturing its products or any of its respective component materials, or the component materials themselves, infringe, or that the use of its products, product candidates or technologies infringe.
Savara or its CMOs or component material suppliers may be exposed to, or threatened with, litigation by third parties alleging that Savara’s products, product candidates and/or technologies infringe its patents and/or other intellectual property rights, or that one or more of the processes for manufacturing its products or any of its respective component materials, or the component materials themselves, or the use of its products, product candidates or technologies, infringe its patents and/or other intellectual property rights. If a third-party patent or other intellectual property right is found to cover its products, product candidates, technologies or its uses, or any of the underlying manufacturing processes or components, Savara could be required to pay damages and could be unable to commercialize its products or use its technologies or methods unless Savara is able to obtain a license to the patent or intellectual property right. A license may not be available to Savara in a timely manner or on acceptable terms, or at all. In addition, during litigation, the third-party alleging infringement could obtain a preliminary injunction or other equitable remedy that could prohibit Savara from making, using, selling or importing its products, technologies or methods.
64
Table of Contents
There generally is a substantial amount of litigation involving patent and other intellectual property rights in the industries in which Savara operates and the cost of such litigation may be considerable. Savara can provide no assurance that its product candidates or technologies will not infringe patents or rights owned by others, licenses to which might not be available to Savara in a timely manner or on acceptable terms, or at all. If a third party claims that Savara or its CMOs or component material suppliers infringe its intellectual property rights, Savara may face a number of issues, including, but not limited to:
| • | infringement and other intellectual property claims which, with or without merit, may be expensive and time consuming to litigate and may divert management’s time and attention from Savara’s core business; |
| • | substantial damages for infringement, including the potential for treble damages and attorneys’ fees, which Savara may have to pay if it is determined that the product and/or its use at issue infringes or violates the third party’s rights; |
| • | a court prohibiting Savara from selling or licensing the product unless the third-party licenses its intellectual property rights to Savara, which it may not be required to do; |
| • | if a license is available from the third party, Savara may have to pay substantial royalties, fees and/or grant cross-licenses to the third party; and |
| • | redesigning Savara’s products or processes so they do not infringe, which may not be possible or may require substantial expense and time. |
No assurance can be given that patents do not exist, have not been filed, or could not be filed or issued, which contain claims covering Savara’s products, product candidates or technology or those of its CMOs or component material suppliers or the use of its products, product candidates or technologies. Because of the large number of patents issued and patent applications filed in the industries in which Savara operates, there is a risk that third parties may allege they have patent rights encompassing Savara’s products, product candidates or technologies, or those of its CMOs or component material suppliers, or uses of its products, product candidates or technologies.
In the future, it may be necessary for Savara to enforce its proprietary rights, or to determine the scope, validity and unenforceability of other parties’ proprietary rights, through litigation or other dispute proceedings, which may be costly, and to the extent Savara is unsuccessful, adversely affect its rights. In these proceedings, a court or administrative body could determine that its claims, including those related to enforcing patent rights, are not valid or that an alleged infringer has not infringed its rights. The uncertainty resulting from the mere institution and continuation of any patent- or other proprietary rights-related litigation or interference proceeding could have a material and adverse effect on its business prospects, operating results and financial condition.
Risks Related to Savara’s Industry
Savara expects competition in the marketplace for its product candidates, should any of them receive regulatory approval.
AeroVanc and Molgradex have received Orphan Drug Designation from FDA and Molgradex has received Orphan Drug Designation from the European Medicines Agency. Orphan Drug Designation will provide market exclusivity in U.S. for seven years and 10 years in Europe, but only if (1) AeroVanc and Molgradex receive market approval before a competitor using the same active compound for the same indication, (2) Savara is able produce sufficient supply to meet demand in the marketplace, and (3) another product with the same active ingredient is not deemed clinically superior. AeroVanc has also received Qualified Infectious Disease Product (QIDP) status extending market exclusivity by an additional five years in addition to any other exclusivity obtained in the United States
The industries in which Savara operates (biopharmaceutical, specialty pharmaceutical, biotechnology and pharmaceutical) are highly competitive and subject to rapid and significant change. Developments by others may
65
Table of Contents
render potential application of any of its product candidates in a particular indication obsolete or noncompetitive, even prior to completion of its development and approval for that indication. If successfully developed and approved, Savara expects its product candidates will face competition. Savara may not be able to compete successfully against organizations with competitive products, particularly large pharmaceutical companies. Many of its potential competitors have significantly greater financial, technical and human resources than Savara, and may be better equipped to develop, manufacture, market and distribute products. Many of these companies operate large, well-funded research, development and commercialization programs, have extensive experience in nonclinical and clinical studies, obtaining FDA and other regulatory approvals and manufacturing and marketing products, and have multiple products that have been approved or are in late-stage development. These advantages may enable them to receive approval from the FDA or any foreign regulatory agency before Savara and prevent Savara from competing due to their orphan drug protections. Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical and biotechnology companies. Furthermore, heightened awareness on the part of academic institutions, government agencies and other public and private research organizations of the potential commercial value of their inventions have led them to actively seek to commercialize the technologies they develop, which increases competition for investment in Savara’s programs. Competitive products may be more effective, easier to dose, or more effectively marketed and sold, than theirs, which would have a material adverse effect on Savara’s ability to generate revenue.
Savara is subject to uncertainty relating to healthcare reform measures and reimbursement policies that, if not favorable to its products, could hinder or prevent its products’ commercial success, if any of its product candidates are approved.
The unavailability or inadequacy of third-party payer coverage and reimbursement could negatively affect the market acceptance of its product candidates and the future revenues Savara may expect to receive from those products. The commercial success of its product candidates, if approved, will depend in part on the extent to which the costs of such products will be covered by third-party payers, such as government health programs, commercial insurance and other organizations. Third-party payers are increasingly challenging the prices and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. These challenges to prices may be problematic to Savara since its products are targeted for a small number of patients (those suffering from orphan diseases) thus requiring Savara to charge very high prices in order to recover development costs and achieve a profit on its revenue. If these third-party payers do not consider its products to be cost-effective compared to other therapies, Savara may not obtain coverage for its products after approval as a benefit under the third-party payers’ plans or, even if Savara does, the level of coverage or payment may not be sufficient to allow Savara to sell its products on a profitable basis.
Significant uncertainty exists as to the reimbursement status for newly approved drug products, including coding, coverage and payment. There is no uniform policy requirement for coverage and reimbursement for drug products among third-party payers in the United States, therefore coverage and reimbursement for drug products can differ significantly from payer to payer. The coverage determination process is often a time-consuming and costly process that will require Savara to provide scientific and clinical support for the use of its products to each payer separately, with no assurance that coverage and adequate payment will be applied consistently or obtained. The process for determining whether a payer will cover and how much it will reimburse a product may be separate from the process of seeking approval of the product or for setting the price of the product. Even if reimbursement is provided, market acceptance of its products may be adversely affected if the amount of payment for its products proves to be unprofitable for healthcare providers or less profitable than alternative treatments or if administrative burdens make its products less desirable to use. Third-party payer reimbursement to providers of its products, if approved, may be subject to a bundled payment that also includes the procedure of administering its products or third-party payers may require providers to perform additional patient testing to justify the use of its products. To the extent there is no separate payment for its product(s), there may be further uncertainty as to the adequacy of reimbursement amounts.
66
Table of Contents
The continuing efforts of governments, private insurance companies, and other organizations to contain or reduce costs of healthcare may adversely affect:
| • | Savara’s ability to set an appropriate price for its products; |
| • | the rate and scope of adoption of its products by healthcare providers; |
| • | its ability to generate revenue or achieve or maintain profitability; |
| • | the future revenue and profitability of its potential customers, suppliers and collaborators; and |
| • | its access to additional capital. |
Savara’s ability to successfully commercialize its products will depend in part on the extent to which governmental authorities, private health insurers and other organizations establish what Savara believes are appropriate coverage and reimbursement for its products. The containment of healthcare costs has become a priority of federal and state governments worldwide and the prices of drug products have been a focus in this effort. For example, there have been several recent U.S. Congressional inquiries and proposed bills designed to, among other things, bring more transparency to drug pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs and the new US President has stated that reducing drug pricing is a priority for his administration. Savara expect that federal, state and local governments in the United States, as well as in other countries, will continue to consider legislation directed at lowering the total cost of healthcare. In addition, in certain foreign markets, the pricing of drug products is subject to government control and reimbursement may in some cases be unavailable or insufficient. It is uncertain whether and how future legislation, whether domestic or abroad, could affect prospects for its product candidates or what actions federal, state, or private payers for healthcare treatment and services may take in response to any such healthcare reform proposals or legislation. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures reforms may prevent or limit its ability to generate revenue, attain profitability or commercialize its product candidates, especially in light of Savara’s plans to price its product candidates at a high level.
Furthermore, Savara expects that healthcare reform measures that may be adopted in the future, including the possible repeal and replacement of the Affordable Care Act, which the Trump administration has stated is a priority, are unpredictable, and the potential impact on its operations and financial position is uncertain, but may result in more rigorous coverage criteria, lower reimbursement, and additional downward pressure on the price Savara may receive for approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payers. The implementation of cost containment measures or other healthcare reforms may prevent Savara from being able to generate revenue, attain profitability or commercialize its products, if approved.
Savara faces potential product liability exposure and, if successful claims are brought against it, Savara may incur substantial liability for a product or product candidate and may have to limit its commercialization. In the future, Savara anticipates that it will need to obtain additional or increased product liability insurance coverage and it is uncertain whether such increased or additional insurance coverage can be obtained on commercially reasonable terms, if at all.
Savara’s business (in particular, the use of its product candidates in clinical studies and the sale of any products for which it obtains marketing approval) will expose Savara to product liability risks. Product liability claims might be brought against Savara by patients, healthcare providers, pharmaceutical companies or others selling or involved in the use of its products. If Savara cannot successfully defend themselves against any such claims, Savara will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for its products and loss of revenue; |
| • | impairment of its business reputation; |
67
Table of Contents
| • | delays in enrolling patients to participate in its clinical studies; |
| • | withdrawal of clinical study participants; |
| • | a “clinical hold,” suspension or termination of a clinical study or amendments to a study design; |
| • | significant costs of related litigation; |
| • | substantial monetary awards to patients or other claimants; and |
| • | the inability to commercialize its products and product candidates. |
Savara maintains limited product liability insurance for its clinical studies, but its insurance coverage may not reimburse Savara or may not be sufficient to reimburse Savara for all expenses or losses it may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, Savara may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect it against losses.
Savara expects that it will expand its insurance coverage to include the sale of commercial products if it obtains marketing approval for any of its product candidates, but Savara may be unable to obtain product liability insurance on commercially acceptable terms or may not be able to maintain such insurance at a reasonable cost or in sufficient amounts to protect Savara against potential losses. Large judgments have been awarded in class action lawsuits based on drug products that had unanticipated side effects. A successful product liability claim or series of claims brought against Savara, if judgments exceed its insurance coverage, could decrease its cash and adversely affect its business.
Risks Related to the Company Following the Merger
The stock price of the Company is expected to be volatile, and the market price of its common stock may drop following the merger.
The market price of the Company’s common stock following the merger could be subject to significant fluctuations following the merger. Market prices for securities of early-stage pharmaceutical, biotechnology and other life sciences companies have historically been particularly volatile. Some of the factors that may cause the market price of the common stock of the Company to fluctuate include:
| • | the ability of the Company to obtain regulatory approvals for its product candidates, and delays or failures to obtain such approvals; |
| • | failure of any of the Company’s product candidates, if approved, to achieve commercial success; |
| • | failure to maintain its existing third party license and supply agreements; |
| • | failure by the Company or its licensors to prosecute, maintain, or enforce its intellectual property rights; |
| • | changes in laws or regulations applicable to its product candidates; |
| • | any inability to obtain adequate supply of its product candidates or the inability to do so at acceptable prices; |
| • | adverse regulatory authority decisions; |
| • | introduction of new products, services, or technologies by its competitors; |
68
Table of Contents
| • | failure to meet or exceed financial and development projections the combined company may provide to the public; |
| • | failure to meet or exceed the financial and development projections of the investment community; |
| • | if securities or industry analysts do not publish research or reports about its business, or if they issue an adverse or misleading opinions regarding its business and stock; |
| • | the perception of the pharmaceutical industry by the public, legislatures, regulators, and the investment community; |
| • | announcements of significant acquisitions, strategic partnerships, joint ventures, or capital commitments by the combined company or its competitors; |
| • | disputes or other developments relating to proprietary rights, including patents, litigation matters, and its ability to obtain patent protection for its technologies; |
| • | additions or departures of key personnel; |
| • | significant lawsuits, including patent or stockholder litigation; |
| • | changes in the market valuations of similar companies; |
| • | general market or macroeconomic conditions; |
| • | sales of its common stock by the combined company or its stockholders in the future; |
| • | trading volume of its common stock; |
| • | announcements by commercial partners or competitors of new commercial products, clinical progress or the lack thereof, significant contracts, commercial relationships or capital commitments; |
| • | adverse publicity relating to the cystic fibrosis market generally, including with respect to other products and potential products in such markets; |
| • | the introduction of technological innovations or new therapies that compete with potential products of the combined organization; |
| • | changes in the structure of health care payment systems; and |
| • | period-to-period fluctuations in the Company’s financial results. |
Moreover, the stock markets in general have experienced substantial volatility that has often been unrelated to the operating performance of individual companies. These broad market fluctuations may also adversely affect the trading price of the Company’s common stock.
In the past, following periods of volatility in the market price of a company’s securities, stockholders have often instituted class action securities litigation against those companies. Such litigation, if instituted, could result in substantial costs and diversion of management attention and resources, which could significantly harm the Company’s profitability and reputation.
The Company will incur costs and demands upon management as a result of complying with the laws and regulations affecting public companies.
The Company will incur significant legal, accounting and other expenses that Savara did not incur as a private company, including costs associated with public company reporting requirements. The Company will also incur costs associated with corporate governance requirements, including requirements under the Sarbanes-Oxley Act, as well as new rules implemented by the SEC and Nasdaq. These rules and regulations are expected to increase the Company’s legal and financial compliance costs and to make some activities more time-consuming and costly. For example, the Company’s
69
Table of Contents
management team will consist of certain officers of Savara prior to the merger, some of whom have not previously managed and operated a public company. These officers and other personnel will need to devote substantial time to gaining expertise regarding operations as a public company and compliance with applicable laws and regulations. These rules and regulations may also make it difficult and expensive for the Company to obtain directors’ and officers’ liability insurance. As a result, it may be more difficult for the Company to attract and retain qualified individuals to serve on the Company’s board of directors or as executive officers of the Company, which may adversely affect investor confidence in the Company and could cause the Company’s business or stock price to suffer.
The Company does not expect to pay any cash dividends in the foreseeable future.
The Company expects to retain its future earnings to fund the development and growth of the combined organization’s business. As a result, capital appreciation, if any, of the common stock of the Company will be stockholders’ sole source of gain, if any, for the foreseeable future.
Future sales of shares by existing stockholders could cause the Company’s stock price to decline.
If stockholders of the Company sell, or indicate an intention to sell, substantial amounts of the Company’s common stock in the public market after legal restrictions on resale and the lock-up agreements discussed in this Current Report on Form 8-K lapse, the trading price of the Company could decline. As of April 27, 2017 immediately following the closing of the merger, the Company had approximately 15.1 million shares of common stock outstanding. Substantially all of such shares of common stock may be sold in the public market; however, approximately 10.5 million of such shares are subject to lock-up restrictions, which restrictions expire beginning on October 27, 2017. If substantial additional shares are sold, or if it is perceived that they will be sold, in the public market, the trading price of the Company’s common stock could decline.
Because the merger likely has resulted in an ownership change under Section 382 of the Code for the Company, the pre-merger net operating loss carryforwards and certain other tax attributes of the Company will be subject to limitation. The net operating loss carryforwards and certain other tax attributes of Savara and of the Company may also be subject to limitations as a result of ownership changes.
If a corporation undergoes an “ownership change” within the meaning of Section 382 of the Code, the corporation’s net operating loss carryforwards and certain other tax attributes arising from before the ownership change are subject to limitations on use after the ownership change. In general, an ownership change occurs if there is a cumulative change in the corporation’s equity ownership by certain stockholders that exceeds fifty percentage points over a rolling three-year period. Similar rules may apply under state tax laws. The merger likely has resulted in an ownership change for the Company and, accordingly, the net operating loss carryforwards and certain other tax attributes of the Company with respect to the pre-closing period will be subject to limitations on use after the merger. The merger may also have resulted in an ownership change for Savara, in which case, Savara’s net operating loss carryforwards and certain other tax attributes would also be subject to limitations. Additional ownership changes in the future could result in additional limitations on the Company’s net operating loss carryforwards. Consequently, even if the Company achieves profitability, it may not be able to utilize a material portion of its net operating loss carryforwards and other tax attributes, which could have a material adverse effect on cash flow and results of operations.
70
Table of Contents
SELECTED HISTORICAL CONSOLIDATED FINANCIAL DATA OF SAVARA
The selected consolidated statements of operations data for the years ended December 31, 2016 and 2015 and the selected consolidated balance sheet data as of December 31, 2016 and 2015 are derived from Savara’s audited consolidated financial statements included elsewhere in this Current Report on Form 8-K. Savara’s historical results are not necessarily indicative of the results that may be expected in any future period.
The selected historical consolidated financial data below should be read in conjunction with the section titled “Savara Management’s Discussion and Analysis of Financial Condition and Results of Operations,” “Risk Factors—Risks Related to Savara’s Capital Requirements and Financial Condition” and Savara’s consolidated financial statements and related notes included elsewhere in this Current Report on Form 8-K.
Consolidated Statements of Operations Data:
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| (in thousands) | ||||||||
| Grant Revenue |
$ | 400 | $ | 54 | ||||
| Operating expenses: |
||||||||
| Research and development |
8,182 | 4,321 | ||||||
| General and administrative |
2,820 | 1,650 | ||||||
| Depreciation |
346 | 6 | ||||||
|
|
|
|
|
|||||
| Total operating expenses |
11,348 | 5,977 | ||||||
|
|
|
|
|
|||||
| Loss from operations |
(10,948 | ) | (5,923 | ) | ||||
|
|
|
|
|
|||||
| Other expense |
332 | 3,076 | ||||||
| Income tax (benefit) |
(357 | ) | — | |||||
|
|
|
|
|
|||||
| Net loss |
$ | (10,923 | ) | $ | (8,999 | ) | ||
|
|
|
|
|
|||||
| Net loss per common share, basic and diluted |
$ | (3.29 | ) | $ | (5.55 | ) | ||
|
|
|
|
|
|||||
| Weighted average shares used in computing net loss per common share, basic and diluted |
3,348,647 | 1,653,259 | ||||||
|
|
|
|
|
|||||
Consolidated Balance Sheet Data:
| As of December 31, | ||||||||
| 2016 | 2015 | |||||||
| (in thousands) | ||||||||
| Cash |
$ | 13,373 | $ | 16,683 | ||||
| Working capital |
11,158 | 15,680 | ||||||
| Total assets |
28,934 | 17,854 | ||||||
| Convertible promissory notes |
3,448 | — | ||||||
| Accumulated deficit |
(38,406 | ) | (27,483 | ) | ||||
| Total stockholders’ equity/(deficit) |
(35,875 | ) | (27,328 | ) | ||||
71
Table of Contents
SAVARA MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND
RESULTS OF OPERATIONS
You should read the following discussion and analysis of Savara’s financial condition and results of operations together with the section entitled “Selected Financial Data” and Savara’s consolidated financial statements and related notes included elsewhere in this Current Report on Form 8-K. This discussion and other parts of this Current Report on Form 8-K contain forward-looking statements that involve risks and uncertainties, such as its plans, objectives, expectations, intentions and beliefs. Savara’s actual results could differ materially from those discussed in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those identified below and those discussed in the section entitled “Risk Factors” included elsewhere in this Current Report on Form 8-K.
Overview
Savara is a clinical stage specialty pharmaceutical company focused on the development and commercialization of product candidates for patients with rare respiratory diseases, including cystic fibrosis (CF), and pulmonary alveolar proteinosis (PAP). Savara’s lead clinical stage product candidate, AeroVanc, is an inhaled formulation of vancomycin, intended for the treatment of persistent methicillin-resistant Staphylococcus aureus (MRSA) lung infection in CF patients. Savara’s second clinical stage product candidate, Molgradex, is an inhaled formulation of recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF), intended for the treatment of PAP. Savara was formed as a corporation in Delaware in 2007. Savara operates in one segment and has its principal offices in Austin, Texas. Since inception, Savara has devoted substantially all of its efforts and resources to identifying and developing its product candidates, recruiting personnel, and raising capital. Savara has incurred operating losses and negative cash flow from operations and has no material product revenue from inception to date. Savara has not yet commenced commercial operations. From inception to December 31, 2016, Savara has raised net cash proceeds of approximately $42.9 million, primarily from private placements of convertible preferred stock and bridge financings.
Savara has never been profitable and has incurred operating losses in each year since inception. Savara’s net losses were $10.9 million, $9.0 million and $6.3 million for the years ended December 31, 2016, 2015 and 2014, respectively. As of December 31, 2016, Savara had an accumulated deficit of $38.4 million. Substantially all of Savara’s operating losses resulted from expenses incurred in connection with its research and development programs and from general and administrative costs associated with its operations.
Savara has chosen to operate by outsourcing its manufacturing and most of its clinical operations. Savara expects to incur significant additional expenses and increasing operating losses for at least the next several years as it initiates and continues the clinical development of, and seeks regulatory approval for, its product candidates and adds personnel necessary to operate as a public company with an advanced clinical candidate pipeline of products. In addition, operating as a publicly traded company would involve the hiring of additional financial and other personnel, upgrading its financial information systems and incurring costs associated with operating as a public company. Savara expects that its operating losses will fluctuate significantly from quarter to quarter and year to year due to timing of clinical development programs and efforts to achieve regulatory approval.
As of December 31, 2016, Savara had cash of $13.4 million. Savara will continue to require substantial additional capital to continue its clinical development and potential commercialization activities. Accordingly, Savara will need to raise substantial additional capital to continue to fund its operations. The amount and timing of its future funding requirements will depend on many factors, including the pace and results of its clinical development efforts. Failure to raise capital as and when needed, on favorable terms or at all, would have a negative impact on its financial condition and its ability to develop its product candidates.
72
Table of Contents
Financial Operations Overview
Research and Development Expenses
Research and development expenses represent costs incurred to conduct research and development, such as the development of Savara’s product candidates. Savara recognizes all research and development costs as they are incurred. Research and development expenses consist primarily of the following:
| • | expenses incurred under agreements with consultants and clinical trial sites that conduct research and development activities on Savara’s behalf; |
| • | laboratory and vendor expenses related to the execution of clinical trials; |
| • | contract manufacturing expenses, primarily for the production of clinical supplies; and |
| • | internal costs that are associated with activities performed by Savara’s research and development organization and generally benefit multiple programs. Where appropriate, these costs are allocated by product candidate. Unallocated internal research and development costs consist primarily of: |
| • | personnel costs, which include salaries, benefits and stock-based compensation expense; |
| • | allocated facilities and other expenses, which include expenses for rent and maintenance of facilities and depreciation expense; and |
| • | regulatory expenses and technology license fees related to development activities. |
The largest component of Savara’s operating expenses has historically been its investment in research and development activities. The following table shows Savara’s research and development expenses for the years ended December 31, 2016, 2015 and 2014:
| Year Ended December 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| (in thousands) | ||||||||||||
| Product candidates: |
||||||||||||
| AeroVanc |
$ | 5,673 | $ | 4,321 | $ | 5,383 | ||||||
| Molgradex |
2,509 | — | — | |||||||||
| Other clinical programs and research related costs |
— | — | 46 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total research and development expenses |
$ | 8,182 | $ | 4,321 | $ | 5,429 | ||||||
|
|
|
|
|
|
|
|||||||
Savara expects research and development expenses will increase in the future as Savara advances its product candidates into and through clinical trials and pursues regulatory approvals, which will require a significant increased investment in regulatory support and contract manufacturing and inventory build-up related costs. In addition, Savara continues to evaluate opportunities to acquire or in-license other product candidates and technologies, which may result in higher research and development expenses due to license fee and/or milestone payments.
73
Table of Contents
The process of conducting clinical trials necessary to obtain regulatory approval is costly and time consuming. Savara may never succeed in timely developing and achieving regulatory approval for its product candidates. The probability of success of Savara’s product candidates may be affected by numerous factors, including clinical data, competition, intellectual property rights, manufacturing capability and commercial viability. As a result, Savara is unable to accurately determine the duration and completion costs of Savara’s development projects or when and to what extent Savara will generate revenue from the commercialization and sale of any of its product candidates.
General and Administrative Expenses
General and administrative expenses consist of personnel costs, facility expenses and expenses for outside professional services, including legal, audit and accounting services. Personnel costs consist of salaries, benefits and stock-based compensation. Facility expenses consist of rent and other related costs. General and administrative costs also include depreciation expense and other supplies. Savara expects to incur additional expenses as a result of becoming a public company following completion of the merger, including expenses related to compliance with the rules and regulations of the SEC and a national securities exchange, additional insurance, investor relations and other administrative expenses and professional services.
Critical Accounting Policies and Estimates
Savara’s management’s discussion and analysis of financial condition and results of operations is based on its consolidated financial statements, which have been prepared in accordance with GAAP. The preparation of these financial statements requires Savara to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. On an ongoing basis, Savara evaluates these estimates and judgments. Savara bases its estimates on historical experience and on various assumptions that Savara believes to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities and the recording of expenses that are not readily apparent from other sources. Actual results may differ materially from these estimates. Savara believes that the accounting policies discussed below are critical to understanding its historical and future performance, as these policies relate to the more significant areas involving management’s judgments and estimates.
Accrued Research and Development Expenses
Savara records accrued expenses for estimated costs of its research and development activities conducted by external service providers, which include the conduct of clinical trial and contract formulation and manufacturing activities. Savara records the estimated costs of development activities based upon the estimated amount of services provided but not yet invoiced, and includes these costs in accrued liabilities in the consolidated balance sheet and within development expense in the consolidated statement of operations and comprehensive loss. These costs are a significant component of Savara’s research and development expenses. Savara records accrued expenses for these costs based on the estimated amount of work completed and in accordance with agreements established with these external service providers.
Savara estimates the amount of work completed through discussions with internal personnel and external service providers as to the progress or stage of completion of the services and the agreed-upon fee to be paid for such services. Savara makes significant judgments and estimates in determining the accrued balance in each reporting period. As actual costs become known, Savara adjusts their accrued estimates.
Stock-based Compensation
Savara recognizes stock-based awards to employees and directors, including stock options, based on the fair value on the grant date using the Black-Scholes option pricing model. The related stock-based compensation is recognized as expense on a straight line-basis over the employee’s or director’s requisite service period
74
Table of Contents
(generally the vesting period). Noncash stock compensation expense is based on awards ultimately expected to vest and is reduced by an estimate for future forfeitures. Forfeitures are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from estimates.
Savara accounts for stock-based compensation arrangements with non-employees using a fair value approach. The fair value of options granted to non-employees is measured using the Black-Scholes option pricing model reflecting similar assumptions for employees except that the expected term is based on the options’ remaining contractual term instead of the simplified method in each of the reported periods. The compensation costs of these arrangements are subject to remeasurement over the vesting terms as earned.
In determining the fair value of the stock-based awards, Savara uses the Black-Scholes option-pricing model and assumptions discussed below. Each of these inputs is subjective and generally requires significant judgment to determine.
Fair Value of Common Stock. The fair value of the shares of common stock underlying stock options has historically been determined by Savara’s board of directors. In order to determine the fair value of the common stock at the time of grant of the option, the Savara Board considered, among other things, valuations performed by an independent third-party. Because there has been no public market for Savara’s common stock, the Savara Board exercised reasonable judgment and considered a number of objective and subjective factors to determine the best estimate of the fair value of Savara’s common stock, including important developments in Savara’s operations, sales of convertible preferred stock, actual operating results and financial performance, the conditions in the life sciences industry and the economy in general, the stock price performance and volatility of comparable public companies, and the lack of liquidity of its common stock, among other factors.
Expected Term. Savara’s expected term represents the period that their stock-based awards are expected to be outstanding and is determined using the simplified method (based on the mid-point between the vesting date and the end of the contractual term) for employee options and the contractual term for non-employee options.
Expected Volatility. Since Savara is privately held and does not have any trading history for its common stock, the expected volatility was estimated based on the average volatility for comparable publicly traded biotechnology companies over a period equal to the expected term of the stock option grants. The comparable companies were chosen based on their similar size, or stage in the life cycle.
Risk-Free Interest Rate. The risk-free interest rate is based on the U.S. Treasury zero coupon issues in effect at the time of grant for periods corresponding with the expected term of option.
Expected Dividend. Savara has never paid dividends on its common stock and has no plans to pay dividends on its common stock. Therefore, Savara used an expected dividend yield of zero.
For the years ended December 31, 2016 , 2015 and 2014, stock-based compensation expense was $209,000, $153,000 and $141,000, respectively. As of December 31, 2016, Savara had $1.2 million of total unrecognized stock-based compensation costs, net of estimated forfeitures, which it expects to recognize over a weighted-average period of 8.5 years.
75
Table of Contents
Results of Operations — Comparison of Years Ended December 31, 2016 and 2015
| Year Ended December 31, |
Dollar | |||||||||||
| 2016 | 2015 | Change | ||||||||||
| (in thousands) | ||||||||||||
| Grant revenue |
$ | 400 | $ | 54 | $ | 346 | ||||||
| Operating expenses: |
||||||||||||
| Research and development |
$ | 8,182 | $ | 4,321 | $ | 3,861 | ||||||
| General and administrative |
2,820 | 1,650 | 1,170 | |||||||||
| Depreciation |
346 | 6 | 340 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
11,348 | 5,977 | 5,371 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(10,948 | ) | (5,923 | ) | (5,025 | ) | ||||||
| Other expense |
332 | 3,076 | (2,744 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Net loss before income taxes |
(11,280 | ) | (8,999 | ) | (2,281 | ) | ||||||
| Income tax benefit |
357 | — | 357 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (10,923 | ) | $ | (8,999 | ) | $ | (1,924 | ) | |||
|
|
|
|
|
|
|
|||||||
Research and development
Research and development expenses increased by $3.9 million, or 89%, to $8.2 million for the year ended December 31, 2016 from $4.3 million for the year ended December 31, 2015. The increase was primarily due to $2.5 million in increased development costs associated with the acquisition of Serendex and the costs associated with Savara’s development of Molgradex. Additionally, research and development increased by approximately $1.3 million related to AeroVanc which is preparing to initiate a Phase 3 study that will begin in 2017. The majority of the AeroVanc increase related to CMC Activity Associated with the Planned Phase 3.
General and administrative
General and administrative expenses increased by $1.2 million, or 71%, to $2.8 million for the year ended December 31, 2016 from $1.7 million for the year ended December 31, 2015. The increase was due to an increase of $0.4 million in connection with various business development activities, including the acquisition costs of Serendex, $0.3 million in personnel costs related to Serendex, administrative and other related costs, $0.1 million in corporate personnel related costs due to increase in headcount, and $0.3 million in accounting/auditing fees and related services.
Other expense
Other expense decreased by $2.7 million, or 89%, to $0.3 million for the year ended December 31, 2016 from $3.1 million for the year ended December 31, 2015. The decrease was due to no longer recording interest expense and discount accretion of the 2014 Notes Payable as these automatically converted in the fourth quarter of 2015.
Income tax benefit
Income tax benefit in 2016 represents a tax benefit provided by the Danish government in the form of a refundable research credit associated with research and development expenditures of Savara’s subsidiary, ApS. There was no tax benefit in 2015, as the subsidiary was not acquired until 2016.
Results of Operations — Comparison of the Years Ended December 31, 2015 and 2014
| Year Ended December 31, |
Dollar | |||||||||||
| 2015 | 2014 | Change | ||||||||||
| (in thousands) | ||||||||||||
| Grant revenue |
$ | 54 | $ | 1,548 | $ | (1,494 | ) | |||||
| Operating expenses: |
||||||||||||
| Research and development |
$ | 4,321 | $ | 5,429 | $ | (1,108 | ) | |||||
| General and administrative |
1,656 | 1,568 | 88 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
5,977 | 6,997 | (1,020 | ) | ||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(5,923 | ) | (5,449 | ) | 474 | |||||||
| Other expense |
3,076 | 833 | 2,243 | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (8,999 | ) | $ | (6,282 | ) | $ | (2,717 | ) | |||
|
|
|
|
|
|
|
|||||||
Grant Revenue
Grant revenue decreased by $1.5 million, or 97%, to $0.1 million for the year ended December 31, 2015 from $1.5 million for the year ended December 31, 2014. The decrease was due to the fact that the grant revenue was associated with the Phase 2 clinical trial for AeroVanc for which the majority of the activity was completed in 2014.
Research and development
Research and development expenses decreased by $1.1 million, or 20%, to $4.3 million for the year ended December 31, 2015 from $5.4 million for the year ended December 31, 2014. The decrease was due to reduced Phase 2 clinical trial activity in 2015 compared to 2014 in connection with Savara’s AeroVanc program as the majority of the Phase 2 activity was completed in 2014.
General and administrative
General and administrative expenses increased by $0.1 million or 6%, to $1.7 million for the year ended December 31, 2015 from $1.6 million for the year ended December 31, 2014. The increase was the result of increased accounting and audit services in 2015 as compared to 2014.
Other expense
Other expense increased by $2.2 million on 269% to $3.1 million for the year ended December 31, 2015 from $0.8 million for the year ended December 31, 2014. The increase was due to interest expense and accretion of the 2014 Notes Payable, which automatically converted into shares of Savara Series C redeemable convertible preferred stock (Series C Preferred Stock”) in the fourth quarter of 2015.
76
Table of Contents
Liquidity and Capital Resources
Sources of Liquidity
Since inception through December 31, 2016, Savara’s operations have been financed primarily by net cash proceeds of $23.3 million from the sale of its convertible preferred stock and the offering of convertible bridge notes in the amount of $19.6 million. As of December 31, 2016, Savara had $13.4 million in cash and an accumulated deficit of $38.4 million. Savara expects that its research and development and general and administrative expenses will increase, and, as a result, Savara anticipates that it will continue to incur increasing losses in the foreseeable future. Therefore, Savara will need to raise additional capital to fund its operations, which may be through the issuance of additional equity, including in connection with the contemplated merger with Mast, and potentially through borrowings.
Note and Warrant Purchase Agreement
During 2014, Savara borrowed $10 million from several investors under convertible subordinate promissory notes (the “2014 Notes”). On December 3, 2015, the 2014 Notes were converted into Series C Preferred Stock in accordance with the automatic conversion provision of the 2014 Notes. The 2014 Notes had an 8.0% simple interest rate per annum computed on the basis of the actual number of days elapsed and a year of 365 days. All unpaid principal, together with any then accrued but unpaid interest was due and payable on the earliest of (i) December 31, 2015 (the “Maturity Date”), (ii) the closing of a change of control as defined, or (iii) the occurrence of an event of default, as defined. The 2014 Notes were pre-payable only with the written consent of the holders of a majority of the principal amount of the then-outstanding 2014 Notes.
On December 3, 2015, the date of the automatic conversion, the 2014 Notes and separated put option liability were surrendered in exchange for Series C Preferred Stock. The debt contract and separated derivative liability were both subject to extinguishment accounting, and a loss in the amount of $226,000 was recorded in the Savara statement of operations and comprehensive loss. The loss was calculated as the difference between the net book value of the 2014 Notes plus the fair value of the put option immediately prior to the automatic conversion, and the fair value of the Series C Preferred Stock into which the 2014 Notes were converted.
Savara has authorized and is pursing the subscription of up to $15 million, in a 2016 Convertible Promissory Note (the “2016 Note”) financing. The 2016 Note carries an annual simple interest rate of 8.0% and is convertible into certain shares of Savara’s equity dependent upon the earlier of the maturity date of June 30, 2018, a subsequent qualified financing, change of control event, Regulation A offering, an Initial Public Offering (“IPO”). The 2016 Notes were amended such that they automatically convert at a stipulated discount upon the consummation of the proposed merger with Mast. In consideration for the purchase of the 2016 Notes on or prior to August 15, 2016, Savara issued to each investor who purchases a 2016 Note, a warrant to purchase Savara’s Series C Preferred Stock. Each warrant will be exercisable for that number of whole shares equal to the quotient obtained by dividing (a) by (b), where (a) is an amount equal to 15% of the principal amount of 2016 Note issued to the investor and (b) is the Series C Preferred Stock price. The exercise price per share shall be the Series C Preferred Stock price. The warrants will expire five (5) years from the date of issuance, or earlier upon an Acquisition or IPO. The warrants will be exercisable upon the earlier to occur of an Acquisition or an IPO. As of December 31, 2016, the carrying value of the 2016 Note was approximately $3.4 million. These warrants have also been amended such that the contemplated merger with Mast would enable the warrant holder to have the right to exercise the warrant any time during the five year expiration period.
77
Table of Contents
Cash Flows
The following table summarizes Savara’s cash flows for the periods indicated:
| Year Ended December 31, | ||||||||
| 2016 | 2015 | |||||||
| (in thousands) |
||||||||
| Cash used in operating activities |
$ | (8,370 | ) | $ | (4,778 | ) | ||
| Cash used in investing activities |
(8 | ) | — | |||||
| Cash provided by financing activities |
5,117 | 8,773 | ||||||
| Effect of exchange rate changes |
(49 | ) | — | |||||
|
|
|
|
|
|||||
| Net increase/(decrease) in cash |
$ | (3,310 | ) | $ | 3,995 | |||
|
|
|
|
|
|||||
Cash flows from operating activities
Cash used in operating activities for the twelve months ended December 31, 2016 was $8.4 million, consisting of a net loss of $10.9 million, which was partially offset by noncash charges of $1.1 million, mainly comprised of depreciation, accretion of discount to convertible promissory notes and stock-based compensation, and by a net increase in liabilities of $1.5 million. The change in Savara’s net operating assets and liabilities was primarily due to an increase accrued liabilities mostly related to research and development costs for both AeroVanc and Molgradex.
Cash used in operating activities for the twelve months ended December 31, 2015 was $4.8 million, consisting of a net loss of $9.0 million, which was partially offset by noncash charges of $3.2 million, including $1.9 million related to the accretion of discount to convertible promissory notes, and by net changes in operating assets and liabilities of $0.9 million. The change in Savara’s net operating assets and liabilities was due primarily to an increase of $1.0 million in grant award receivables and a decrease in accrued liabilities of $0.2 million related to the decrease in research and development activities.
Cash flows from investing activities
Cash used in investing activities for all periods presented was related to purchases of property and equipment, primarily related to office and computer equipment.
Cash flows from financing activities
Cash provided by financing activities for all periods presented was related to proceeds from the issuance of convertible preferred stock, net of issuance costs and/or issuance of convertible promissory notes.
Future Funding Requirements
Savara has not generated any revenue from product sales. Savara does not know when, or if, it will generate any revenue from product sales. Savara does not expect to generate any revenue from product sales unless and until it obtains regulatory approval for and commercializes any of its product candidates. At the same time, Savara expects its expenses to increase in connection with its ongoing development and manufacturing activities, particularly as Savara continues the research, development, manufacture and clinical trials of, and seeks regulatory approval for, its product candidates. Upon the closing of the merger, Savara expects to incur additional costs associated with operating as a public company. In addition, subject to obtaining regulatory approval of any of its product candidates, Savara anticipates that it will need substantial additional funding in connection with its continuing operations.
78
Table of Contents
As of December 31, 2016, Savara had cash of $13.4 million. Savara will continue to require substantial additional capital to continue its clinical development and potential commercialization activities. Accordingly, Savara will need to raise substantial additional capital to continue to fund its operations. The amount and timing of its future funding requirements will depend on many factors, including the pace and results of its clinical development efforts. Failure to raise capital as and when needed, on favorable terms or at all, would have a negative impact on its financial condition and Savara’s ability to develop its product candidates.
Until Savara can generate a sufficient amount of product revenue to finance its cash requirements, it expects to finance its future cash needs primarily through the issuance of additional equity subsequent to the Merger, and potentially through borrowings, grants and strategic alliances with partner companies. To the extent that Savara raises additional capital through the issuance of additional equity or convertible debt securities, the ownership interest of Savara’s stockholders will be diluted, and the terms of these securities may include liquidation or other preferences that adversely affect the rights of existing stockholders. Debt financing, if available, may involve agreements that include covenants limiting or restricting Savara’s ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If Savara raises additional funds through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, Savara may have to relinquish valuable rights to its technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to Savara. If Savara is unable to raise additional funds through equity or debt financings when needed, Savara may be required to delay, limit, reduce or terminate its product development or commercialization efforts or grant rights to develop and market product candidates to third parties that Savara would otherwise prefer to develop and market itself.
Contractual Obligations and Other Commitments
As of December 31, 2016, Savara leased its office facilities under a non-cancellable operating lease. The lease term was extended for a period of 48 months, commencing on December 1, 2015, and expiring on November 30, 2019. Savara recognizes rent expense on a straight-line basis over the operating lease term. The lease is cancellable three years after execution of the lease if Savara notifies the property owner of its intention to cancel the lease by the end of second year of the lease. The future minimum annual lease payments under the operating lease are as follows (in thousands):
| Year ending December 31, |
||||
| 2017 |
$ | 172 | ||
| 2018 |
174 | |||
| 2019 |
161 | |||
|
|
|
|||
| Total minimum lease payments |
$ | 507 | ||
|
|
|
|||
As of December 31, 2016, Savara leases certain research and development equipment as part of a contract manufacturing arrangement. The leased equipment is accounted for as a capital lease, and the present value of the future minimum lease payments are recorded as a liability on the balance sheet as of December 31, 2016. The future minimum annual lease payments under the capital lease are as follows (in thousands):
| Year ending December 31, |
||||
| 2017 |
$ | 486 | ||
| 2018 |
312 | |||
| 2019 |
313 | |||
|
|
|
|||
| Total minimum lease payments |
1,111 | |||
| Less: imputed interest |
(90 | ) | ||
|
|
|
|||
| Total capital lease obligation |
$ | 1,021 | ||
|
|
|
|||
79
Table of Contents
License and Royalty Agreements
Savara is also subject to certain contingent payments to the Cystic Fibrosis Foundation Therapeutics (CFFT) in connection with a $1.7 million award from the CFFT that was provided to Savara in support of AeroVanc research (CFF Award). A payment is due to the CFFT equal to three (3) times the amount of the CFF Award upon approval of AeroVanc for commercial use. The payment is owed in equal installments of 33% due 60 days after first commercial sale; 33% due 90 days of the first anniversary of the first commercial sale; and 34% due within 90 days of 2nd anniversary of first commercial sale. As Savara’s product has not yet been approved for commercial use, Savara has not recorded a liability for the commercial approval payment.
In addition, if net sales exceed $50.0 million for any calendar year occurring during the first five years after the first commercial sale, Savara must remit payment to the CFFT equal to one (1) times the CFF Award. Furthermore, if net sales exceed $100.0 million for any calendar year occurring during the first five years after first commercial sale, Savara must remit an additional payment to the CFFT equal to one (1) times the CFF Award. Given Savara has not recognized any sales from AeroVanc, Savara has not recorded a liability for any amounts due as additional royalties.
Savara is subject to various manufacturing royalties and payments related to Molgradex. Upon the successful development, registration and attainment of approval by the proper health authorities, such as the FDA, in any territory except Latin America, Central America and Mexico, Savara must pay a royalty of three percent (3%) on annual net sales to the manufacturer of its Active Pharmaceutical Ingredients (“API”). Under this agreement with the API manufacturer, no signing fee or milestones are included in the royalty payments, and there is no minimum royalty. Additionally, Savara has a commitment to acquire a working cell bank and a master cell bank for $1,950,000 from this API manufacture in the third quarter of 2017.
Savara is also subject to certain contingent milestone payments up to approximately seven million euros based upon various development activities and regulatory approvals payable to Savara’s manufacturer of its nebulizer used to administer Molgradex. In addition to these milestones, Savara will owe a royalty to the manufacturer of its nebulizer based on net sales. The royalty rate ranges from three and a half percent (3.5%) to five percent (5%) depending on the device technology used by Savara to administer to product.
Acquisition of Serendex Pharmaceuticals
On July 15, 2016, Savara closed on a Business Transaction Agreement (“BTA”) under which Savara acquired certain assets, liabilities, employees, and subsidiaries of Serendex Pharmaceuticals A/S (“Seller”), a limited liability company incorporated under the laws of Denmark which delisted from the Oslo Axxes (“Oslo Stock Exchange”) on or about May 4, 2016. The Seller’s wholly owned subsidiaries include Pharmaorigin ApS and Drugrecure ApS (the “Subsidiaries”) which are limited liability companies incorporated under the laws of Denmark. The Seller was a biopharmaceutical development company which, directly and through its Subsidiaries, advanced a pipeline and portfolio of novel inhalation therapies and related technologies for the treatment of severe pulmonary conditions. Its primary focus was on the medicinal product Molgradex® (an inhalation formulation of recombinant human GM-CSF for the treatment of pulmonary alveolar proteinosis). The purchase price consists of 3,353,925 shares of Savara’s common stock, subject to a hold back of 670,785 shares of common stock by Savara in the name of the Seller as security for the Seller’s obligations under the BTA until the lapse of the deadline for submission of claims, and $21.5 million of contingent cash consideration based upon the achievement of certain milestones.
Other Contracts
Savara enters into contracts in the normal course of business with various third parties for research studies, clinical trials, testing and other services. These contracts generally provide for termination upon notice, and therefore Savara believes that its non-cancelable obligations under these agreements are not material except for certain obligations under its agreement for its capitalized lease asset.
80
Table of Contents
Off-Balance Sheet Arrangements
Savara has not entered into any off-balance sheet arrangements and do not have any holdings in variable interest entities.
Recent Accounting Pronouncements
In November 2015, the FASB issued Accounting Standards Update 2015-17, “Income Taxes, Balance Sheet Classification of Deferred Taxes” (“ASU 2015-17”), which eliminates the current requirement for reporting entities to present deferred tax liabilities and assets as current and noncurrent in a classified balance sheet. Instead, reporting entities will be required to classify all deferred tax assets and liabilities as noncurrent. This guidance is effective for fiscal years beginning after December 15, 2016. The adoption of this standard is not expected to have a material impact on Savara’s financial statements.
In February 2016, the FASB issued Accounting Standards Update 2016-02, “Leases” (“ASU 2016-02”). The update aims at making leasing activities more transparent and comparable, and requires substantially all leases be recognized by lessees on their balance sheet as a right-of-use asset and a corresponding lease liability, including leases currently accounted for as operating leases. The update also requires improved disclosures to help users of financial statements better understand the amount, timing and uncertainty of cash flows arising from leases. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018 with early adoption permitted. Savara is currently evaluating the impact of the adoption of ASU 2016-02 on its financial statements.
In March 2016, the FASB issued Accounting Standards Update 2016-09, “Compensation — Stock Compensation: Improvements to Employee Share-Based Payment Accounting” (“ASU 2016-09”). ASU 2016-09 changes certain aspects of the accounting for share-based payment awards, including accounting and cash flow classification for excess tax benefits and deficiencies; income tax withholding obligations; forfeitures; and cash flow classification. ASU 2016-09 is effective for Savara for annual periods beginning after December 15, 2017, and interim periods within annual periods beginning after December 15, 2018 with early adoption permitted. Savara is currently evaluating the effect of this new guidance on its financial statements.
In November 2016, the FASB issued ASU 2016-18, Statement of Cash Flows (Topic 230) Restricted Cash. The ASU clarifies the classification and presentation of changes in restricted cash on the statement of cash flows. The ASU is effective for public companies for fiscal years beginning after December 15, 2017, including interim periods within those fiscal years. Savara does not expect the adoption of ASU 2016-18 will have a material impact on its consolidated financial statements.
81
Table of Contents
QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT SAVARA MARKET RISK
As of December 31, 2016, Savara had cash of $13.4 million, which consisted of bank deposits. Such interest-earning instruments carry a degree of interest rate risk; however, historical fluctuations of interest income have not been significant. Savara has not been exposed nor does it anticipate being exposed to material risks due to changes in interest rates. A hypothetical 1% change in interest rates during any of the periods presented would not have had a material impact on Savara’s consolidated financial statements.
Savara has ongoing operations in Denmark as a result of its acquisition of Serendex and pays those vendors in local currency (Danish Krone) or Euros. Savara does not participate in any foreign currency hedging activities and it does not have any other derivative financial instruments. Savara did not recognize any significant exchange rate losses during the nine-month period ended December 31, 2016. A 10% change in the krone-to-dollar or euro-to-dollar exchange rate on December 31, 2016 would not have had a material effect on Savara’s results of operations or financial condition.
82
Table of Contents
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table and the related notes present information on the beneficial ownership of shares of the Company’s capital stock as of April 27, 2017, after giving effect to the Reverse Stock Split and the Merger and related transactions, by:
| • | each of the Company’s directors as of the closing of the merger; |
| • | each of the Company’s executive officers as of the closing of the merger and each of the Company’s named executive officers as reflected in the Company’s Annual Report on form 10-K for the year ended December 31, 2016; |
| • | all of the Company’s current directors and executive officers as a group; and |
| • | each stockholder known by the Company to beneficially own more than five percent of the Company’s common stock. |
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. Shares of common stock that may be acquired by an individual or group within 60 days of April 27, 2017, pursuant to the exercise of options or warrants, are deemed to be outstanding for the purpose of computing the percentage ownership of such individual or group, but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table.
Except as indicated in footnotes to this table, we believe that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them, based on information provided to the Company by such stockholders. Unless otherwise indicated, the address for each stockholder listed is: c/o Savara Inc., 900 S. Capital of Texas Highway, Las Cimas IV, Suite 150, Austin, TX 78746.
| Name and Address of Beneficial Owner |
Shares Beneficially Owned |
Percent of Outstanding |
||||||
| Principal Stockholders: |
||||||||
| Serenova A/S(1) |
1,965,400 | 13.04 | % | |||||
| Directors and Named Executive Officers: |
||||||||
| Robert Neville(2) |
602,469 | 3.94 | % | |||||
| Nevan Elam(3) |
46,434 | * | ||||||
| Richard J. Hawkins(4) |
44,090 | * | ||||||
| Yuri Pikover(5) |
305,398 | 2.02 | % | |||||
| Joseph S. McCracken(6) |
37,144 | * | ||||||
| David Ramsay(7) |
2,570 | * | ||||||
| Matthew Pauls(8) |
650 | * | ||||||
| Taneli Jouhikainen(9) |
361,803 | 2.38 | % | |||||
| David Lowrance(10) |
27,213 | * | ||||||
| Brian Culley(11) |
34,323 | * | ||||||
| Brandi Roberts(12) |
13,255 | * | ||||||
| Edwin Parsley(13) |
12,779 | * | ||||||
| Shana Hood(14) |
6,479 | * | ||||||
| R. Martin Emanuele(15) |
18,502 | * | ||||||
| Gregory D. Gorgas(16) |
25,482 | * | ||||||
| All current executive officers and directors as a group (9 persons)(17) |
1,427,771 | 9.15 | % | |||||
| * | Represents beneficial ownership of less than 1% of the shares of Common Stock. |
| (1) | Includes 393,080 shares held in escrow by the Company until July 19, 2017 pursuant to the terms of the Business Transfer Agreement dated May 13, 2016 between Serendex Pharmaceuticals A/S and Savara Inc. |
| (2) | Includes 224,334 shares issuable upon the exercise of options held by Mr. Neville that are exercisable within 60 days of April 27, 2017. |
| (3) | Consists of 46,434 shares issuable upon the exercise of options held by Mr. Elam that are exercisable within 60 days of April 27, 2017. |
| (4) | Consists of 44,090 shares issuable upon the exercise of options held by Mr. Hawkins that are exercisable within 60 days of April 27, 2017. |
| (5) | Includes 275,445 shares held by 37Ventures, LLC, and Mr. Pikover is managing director of 37Ventures, LLC. Also includes (i) 29,104 shares issuable upon the exercise of options held by Mr. Pikover and (ii) 849 shares issuable upon the exercise of outstanding warrants, in each case that are exercisable within 60 days of April 27, 2017. |
| (6) | Includes (i) 29,104 shares issuable upon the exercise of options held by Dr. McCracken and (ii) 424 shares issuable upon the exercise of outstanding warrants, in each case that are exercisable within 60 days of April 27, 2017. |
| (7) | Includes 1,142 shares issuable upon settlement of RSUs that vested on April 27, 2017. |
| (8) | Consists of 650 shares issuable upon settlement of RSUs that vested on April 27, 2017. |
| (9) | Includes 136,977 shares issuable upon the exercise of options held by Dr. Jouhikainen that are exercisable within 60 days of April 27, 2017. |
| (10) | Consists of 27,213 shares issuable upon the exercise of options held by Mr. Lowrance that are exercisable within 60 days April 27, 2017. |
| (11) | Includes 33,831 shares issuable upon settlement of RSUs that vested on April 27, 2017. |
| (12) | Includes 12,713 shares issuable upon settlement of RSUs that vested on April 27, 2017. |
| (13) | Consists of 12,779 shares issuable upon settlement of RSUs that vested on April 27, 2017. |
| (14) | Consists of 6,479 shares issuable upon settlement of RSUs that vested on April 27, 2017. |
| (15) | Consists of (a) 14,294 shares of common stock subject to options that currently are exercisable and (b) 4,208 outstanding shares of common stock held directly by Dr. Emanuele. The outstanding shares of common stock owned by Dr. Emanuele are subject to the Stockholders’ Voting and Transfer Restriction Agreement, dated February 12, 2011, by and among the Mast Therapeutics, Inc., each of the former principal stockholders of SynthRx, Inc. and, solely with respect to Section 3(c), the Stockholders’ Agent, pursuant to which Dr. Emanuele agreed, with respect to every action or approval by written consent of our stockholders (subject to limited exceptions), to vote these shares in such manner as the Company directs and granted an irrevocable proxy to the Company for the duration of such agreement. |
| (16) | Includes 25,482 shares issuable upon the exercise of options that are currently exercisable. |
| (17) | Includes 612,005 shares held of record by the Company’s directors and executive officers, 537,256 shares issuable upon the exercise of options held by the Company’s directors and executive officers that are exercisable within 60 days of April 27, 2017, 1,273 shares issuable upon the exercise of warrants held by the Company’s directors and executive officers that are exercisable within 60 days of April 27, 2017, 1,792 shares issuable upon settlement of RSUs within 60 days of April 27, 2017 and 275,445 shares held by entities over which the Company’s directors and executive officers may be deemed to have voting and dispositive power. |
83
Table of Contents
Executive Officers and Directors
At the effective time of the merger, each of Robert Neville, Nevan Elam, Richard J. Hawkins, Joseph S. McCracken, Yuri Pikover, Matthew Pauls and David A. Ramsay was appointed to our board of directors and such individuals constitute our board of directors as of the date of this report. Additionally, pursuant to the Merger Agreement, our executive management team changed at the effective time of the merger by the resignation of the then-serving executive officers and the appointment of Robert Neville as our Chairman and Chief Executive Officer, Taneli Jouhikainen as our President and Chief Operating Officer and David Lowrance as our Chief Financial Officer.
The following table sets forth the names, ages and positions of each of our directors and executive officers as of the date of this report:
| Name |
Age | Position(s) | ||||
| Executive Officers |
||||||
| Robert Neville |
51 | Chairman and Chief Executive Officer | ||||
| Taneli Jouhikainen |
50 | President and Chief Operating Officer | ||||
| David Lowrance |
49 | Chief Financial Officer | ||||
| Non-Employee Directors |
||||||
| Nevan Elam |
49 | Director | ||||
| Richard J. Hawkins |
68 | Director | ||||
| Joseph S. McCracken |
64 | Director | ||||
| Yuri Pikover |
55 | Director | ||||
| Matthew Pauls |
46 | Director | ||||
| David A. Ramsay |
52 | Director | ||||
Executive Officers
Robert Neville has served as Savara’s Chairman and CEO since he co-founded the company in June 2008. From January 2003 to December 2004, he served as managing director of Clockwise Consulting, where he worked with the Texas Children’s Hospital and the Baylor College of Medicine to develop an ICU-based monitoring and diagnostic device. From June 2000 to December 2002, he served as Vice President of Engineering at BMC Software, a software company providing information technology management solutions, where he oversaw the integration and expansion of the product line based on the acquisition of his previous company, Evity, Inc. From June 1998 to May 2000, Mr. Neville served as co-founder and CEO of Evity, Inc., a developer of a web-based application that enables customers to measure their transaction performance, where he led the development of the company until its acquisition by BMC Software. Based on his work at Savara and Evity, Mr. Neville was honored as a two-time finalist for the Ernst & Young Entrepreneur of the Year award. Mr. Neville holds a post-graduate Engineering degree from the University of Natal South Africa. The Company believes Mr. Neville’s experience as Chief Executive Officer of Savara and his previous service in executive positions at various companies qualifies him to serve on the Board.
Dr. Taneli Jouhikainen is a co-founder of Savara, and has served as Chief Operating Officer since October 2009 and President since December 2014. From October 2006 until September 2009, he served at Akela Pharma, Inc., a public clinical stage specialty pharmaceutical company focused on orphan drugs and inhalation
84
Table of Contents
products, first as Head of Corporate Development, and subsequently as interim CEO until the company’s merger with Nventa Biopharmaceuticals. From January 2004 to September 2006, he served as President of LAB Pharma Oy, and Head of the Drug Development Business Unit of its parent company, LAB International, Inc., a public clinical stage specialty pharmaceutical company. From January 2001 to January 2004, he served at Focus Inhalation Oy, a clinical stage specialty pharmaceutical company focusing on inhaled products, first as Vice President of Business Development & Strategy, and subsequently as President and Chief Executive Officer, until the merger of Focus Inhalation with LAB International, Inc. From May 1994 to December 2000, he served at Schering AG, a global pharmaceutical company, first as Research Manager, and subsequently as Head of Clinical Development. Dr. Jouhikainen holds an MD and a Ph.D. in hematology and immunology from the University of Helsinki, and an MBA from the Helsinki School of Economics.
David Lowrance has served as Savara’s Chief Financial Officer since November 2016. From September 2014 to October 2016, Mr. Lowrance served as the Chief Financial Officer and Treasurer of Edgemont Pharmaceuticals, a fully-integrated specialty pharmaceutical company with multiple marketed products in the CNS space. From April 2011 to September 2014, Mr. Lowrance served as the Chief Financial Officer and Secretary of Acucela Inc., a clinical-stage biotechnology company that specializes in ophthalmic therapeutics, where he was responsible for overseeing all aspects of Acucela’s day-to-day operations, business development and growth endeavors, investor relations and corporate communications. While at Acucela, Mr. Lowrance helped lead a $162 million international IPO, with a listing on the Tokyo Stock Exchange. From March 2003 to April 2011, Mr. Lowrance was Vice President and Chief Financial Officer of Cumberland Pharmaceuticals Inc., a specialty pharmaceutical company focused on commercializing branded prescription products, where he oversaw all aspects of finance and accounting, business and growth strategy and product development. Mr. Lowrance, a CPA, holds a B.B.A. in Accounting from the University of Georgia.
Non-Employee Directors
Nevan Elam has served as a member of the Savara Board since February 2009. Mr. Elam is currently the President, Chief Executive Officer and Chairman of AntriaBio, Inc., a biopharmaceutical company focused on developing novel extended release therapies. Prior to his tenure at AntiraBio which began in October 2012, Mr. Elam served for three years as the Chief Executive Officer and President of AeroSurgical Ltd., a medical device company operating out of Ireland. Prior to his service with AeroSurgical Ltd., Mr. Elam was Head of the Pulmonary Business Unit and Senior Vice President of Nektar Therapeutics. Earlier in his career he was a founder and Chief Financial Officer of E2open as well as a Partner in the corporate practice of the law firm of WSGR. In addition to serving on the AntriaBio Board of Directors, he also serves on the Board of Directors of pH Pharmaceuticals in Seoul, Korea. Mr. Elam received his Juris Doctorate from Harvard Law School and a Bachelors of Arts from Howard University. The Company believes Mr. Elam’s broad experience with pharmaceutical companies, including advising them of their unique legal and regulatory obligations, qualifies him to serve on the Board.
Richard J. Hawkins has served as a member of the Savara Board since October 2010. Since September 2010, Mr. Hawkins has served as President and Chief Executive Officer of Lumos Pharma, Inc., a clinical stage biotechnology company focused on bringing novel therapies to patients with severe, rare, and genetic diseases, whose medical needs are unmet. From 2000 to 2010, Mr. Hawkins, founded and advised numerous pharmaceutical companies including Sensus, where he served as co-founder and Chairman until being sold to Pfizer. From 1981 to 2000, Mr. Hawkins was founder, President and CEO of Pharmaco and guided the company’s growth to over 2,000 employees. The company later merged with PPD of Wilmington, NC to form PPD Pharmaco, one of the largest clinical contract research organizations in the world. Mr. Hawkins received his Bachelor of Science in Biology from Ohio University. The Company believes Mr. Hawkins’s experience in the pharmaceutical and life sciences industries as well as his broad management experience qualify him to serve on the Board.
85
Table of Contents
Joseph S. McCracken has served as a member of the Savara Board since October 2013 and currently advises biopharmaceutical companies on the design and implementation of corporate strategy and business development initiatives. Joe also serves on the boards of several biopharmaceutical companies, including Alkahest, Inc., Genkyotex S.A., Nexvet Biopharma (NASDAQ: NVET) and Regimmune Inc. From July 2011 to September 2013, Dr. McCracken was Vice President and Global Head of Business Development & Licensing for Roche Pharma, a research-focused healthcare company, where he was responsible for Roche Pharma’s global in-licensing and out-licensing activities. From October 2009 until July 2011 he was General Manager, Roche Pharma Japan & Asia Regional Head, Roche Partnering. Prior to joining Roche Pharma, Dr. McCracken held the position of Vice President, Business Development at Genentech for more than 10 years, and previously held similar positions at Aventis Pharma and Rhone-Poulenc Rorer. Dr. McCracken holds a Bachelor of Science in Microbiology, a Master of Science in Pharmacology and a Doctorate of Veterinary Medicine from The Ohio State University. The Company believes Dr. McCracken’s extensive experience in the biotechnology and pharmaceutical industries qualifies him to serve on the Board.
Yuri Pikover has served as a member of the Savara Board since October 2013. Since 1999 Mr. Pikover served as Managing Director of 37Ventures, a boutique venture fund focusing on growing early-stage startups. From 1999-2002 Mr. Pikover was Chairman and Chief Executive Officer of Access360. From 1993 to 1999, Mr. Pikover was co-founder and Executive Vice President of Xylan and helped lead the fast growing organization going public in 1996 and acquisition by Alcatel in 1999. The Company believes Mr. Pikover’s extensive experience as an investor and board member in pharmaceutical and life sciences companies and his knowledge gained from service on such boards qualify him to be a member of the Board.
Matthew Pauls has served as a member of the Board since October 2015 and has served as chair of the Mast Board since March 2016. Mr. Pauls currently serves as President and Chief Executive Officer of Strongbridge Biopharma plc (NASDAQ: SBBP), a biopharmaceutical company focused on therapies that target rare diseases, a position he has held since August 2014. He also has served as a member of the board of directors of Strongbridge since September 2015. Prior to Strongbridge, from April 2013 to August 2014, Mr. Pauls was Chief Commercial Officer of Insmed, Inc., a publicly traded global biopharmaceutical company focused on rare diseases. Prior to Insmed, Mr. Pauls worked at Shire Pharmaceuticals, a global specialty biopharmaceutical company, from 2007 to April 2013, most recently as Senior Vice President, Head of Global Commercial Operations from May 2012 to April 2013. Earlier in his career, from 1997 to 2007, Mr. Pauls held senior positions at Bristol-Myers Squibb in Brand Management and Payor Marketing and at Johnson & Johnson in various U.S. and global commercial roles. Mr. Pauls holds B.S. and M.B.A. degrees from Central Michigan University and a J.D. from Michigan State University College of Law. Mr. Pauls’ leadership experience and extensive commercialization, strategic planning and operations experience in the biopharmaceutical industry and particularly with therapies for rare diseases qualify him to serve as a member of the Board.
David A. Ramsay has served as member of the Board since June 2011. Mr. Ramsay served as Chief Financial Officer of Halozyme Therapeutics, Inc. (NASDAQ: HALO), a biotechnology company developing and commercializing novel oncology therapies, from May 2013 until his retirement in July 2015 and from 2003 to May 2009. He also served as Halozyme’s Vice President, Corporate Development from May 2009 to May 2013. Mr. Ramsay currently provides consulting services to biotechnology companies, both publicly traded and privately-held. From 2000 to 2003, Mr. Ramsay was Vice President, Chief Financial Officer of Lathian Systems, Inc., a provider of technology-based sales solutions for the life science industry. From 1998 to 2000, he was with Valeant Pharmaceuticals International, Inc. (formerly ICN Pharmaceuticals, Inc.), a multinational specialty pharmaceutical company, where he served as Vice President, Treasurer and Director, Corporate Finance. Mr. Ramsay began his career at Deloitte & Touche, where he obtained his CPA license. Mr. Ramsay holds a B.S. in business administration from the University of California, Berkeley and a M.B.A. with a dual major in finance and strategic management from The Wharton School at the University of Pennsylvania. Mr. Ramsay’s significant experience as chief financial officer of life science companies, particularly his experiences at Halozyme during its successful development and its commercialization of its first products, and at a large audit and financial advisory firm, qualify him to serve on the Board.
86
Table of Contents
Composition of the Board of Directors
The Board currently consists of seven directors, and each director’s term expires upon the election and qualification of successor directors at the annual meeting of the stockholders to be held in 2017.
There are no family relationships among any of the directors and executive officers.
Director Independence
The Board has determined that each of its directors other than Robert Neville is independent as defined under Nasdaq listing standards. The Board has also determined that each current member of the Nominating and Governance Committee is independent as defined under the Nasdaq listing standards, and that each current member of the Audit Committee and Compensation Committee is independent as defined under the Nasdaq listing standards and applicable SEC rules. In making this determination, the Company’s board of directors found that none of these directors had a material or other disqualifying relationship with the Company.
Committees of the Board of Directors
The Board has an Audit Committee, a Compensation Committee and a Nominating and Governance Committee.
Audit Committee
The Audit Committee of the Board was established by the Company’s board of directors in accordance with Section 3(a)(58)(A) of the Exchange Act to oversee the Company’s corporate accounting and financial reporting processes and audits of its financial statements. For this purpose, the Audit Committee performs several functions, including, among other things:
| • | appointing and providing for the compensation of the independent registered public accounting firm to be engaged to prepare and issue an audit report and perform other audit, review or attest services; |
| • | approving any other permissible non-audit services to be provided by the independent auditor; |
87
Table of Contents
| • | overseeing the work and evaluating the performance of the independent auditor, and, if so determined by the audit committee, terminating and replacing the independent auditor; |
| • | reviewing and discussing, including with management and the independent auditor, the annual and quarterly financial statements; |
| • | reviewing any proposed significant changes to accounting principles and practices; |
| • | reviewing any material changes to the system of internal control over financial reporting; |
| • | reviewing management’s report on effectiveness of internal control over financial reporting and, if applicable, the independent auditor’s audit of the effectiveness of Mast’s internal control over financial reporting; |
| • | establishing a procedure for receipt, retention and treatment of any complaints or concerns received by the Company about accounting, internal accounting controls or auditing matters; |
| • | reviewing, approving and overseeing any related party transaction that would require disclosure pursuant to Item 404 of Regulation S-K; |
| • | overseeing the implementation and enforcement of the Company’s insider trading policy; and |
| • | reviewing and evaluating any significant financial risk exposures facing the Company and the steps the Company’s management has taken to control and monitor such exposures. |
The Company’s management has the primary responsibility for its consolidated financial statements and the reporting process including its system of internal accounting and financial controls.
The Audit Committee consists of Mr. Ramsay, who serves as its chairman, Mr. Pikover and Mr. Hawkins. The Board reviews the Nasdaq listing standards definition of independence for Audit Committee members on an annual basis and has determined that all current members of the Audit Committee are independent (as independence is currently defined by listing standards and Rule 10A-3 of the Exchange Act.
The Board has also determined that Mr. Ramsay qualifies as an “audit committee financial expert,” as defined in applicable SEC rules. The Board made a qualitative assessment of Mr. Ramsay’s level of knowledge and experience based on a number of factors, including his formal education and experience in financial roles.
Compensation Committee
The Compensation Committee of the Board acts on behalf of the Board to review, adopt or recommend for adoption, and oversee Company’s compensation strategy, policies, plans and programs. For this purpose, the Compensation Committee performs several functions, including, among other things:
| • | reviewing and recommending to the Board for its determination and approval the amount, form and terms of compensation of the Company’s Chief Executive Officer and other “officers” (as such term is defined under the Nasdaq listing standards); |
88
Table of Contents
| • | reviewing and making recommendations to the Board regarding the Company’s overall compensation strategy and policies; |
| • | reviewing and making recommendations regarding the Company’s equity and/or cash incentive plans and other benefit plans and, to the extent as may be permitted or required under such plans, the committee has the power and authority to administer the plans, establishes guidelines, interpret plan documents, select participants, and approve grants and awards thereunder; |
| • | granting equity awards to non-officer employees and consultants in accordance with the terms of the Company’s equity incentive plan and to establish compensation policies and practices applicable to non-officer employees; |
| • | evaluating the relationship between executive officer compensation policies and practices and corporate risk management to confirm those policies and practices do not incentivize excessive risk-taking; |
| • | evaluating and making recommendations to the Board regarding the compensation of non-employee directors; |
| • | retaining, obtaining the advice of, engaging, compensating and terminating compensation consultants, legal counsel and such other advisors as it deems necessary and advisable to assist it in carrying out its responsibilities and functions; and |
| • | appointing, compensating and overseeing the work of any of its compensation consultants, legal counsel and other advisors. |
The Compensation Committee consists of Mr. Elam who serves as its chairman, Dr. McCracken and Mr. Pauls. All members of the Compensation Committee are independent as independence is currently defined under the Nasdaq listing standards and Rule 10C-1 of the Exchange Act.
Nominating and Governance Committee
The Nominating and Governance Committee of the Board is responsible for identifying, reviewing and evaluating candidates to serve as directors of the Company (consistent with criteria approved by the Board), reviewing and evaluating incumbent directors, selecting or recommending to the Board for selection candidates for election to the Board, making recommendations to the Board regarding the membership of the committees of the Board, assessing the performance of the Board, and developing a set of corporate governance principles for the Company. The responsibilities of the Nominating and Governance Committee relating to the nomination of directors include, among other things, the following:
| • | identifying and recommending to the Board nominees for possible election to the Company’s board of directors; |
| • | evaluating and making recommendations to the Board regarding its size, composition and leadership structure; |
| • | reviewing and assessing the Company’s corporate governance guidelines and recommending any proposed changes thereto to the Board; |
89
Table of Contents
| • | reviewing and making recommendations to the Board regarding issues of executive officer succession planning and providing oversight with respect to corporate governance matters. |
In recommending candidates for appointment or election to the Board, the Nominating and Governance Committee considers the appropriate balance of experience, skills and characteristics required of the Board and seeks to insure that at least a majority of the directors are independent under Nasdaq listing standards and that the Board’s Audit Committee and Compensation Committee will be comprised of directors who meet applicable Nasdaq listing standards and SEC rules regarding qualifications to serve on such committees. Candidates for director are selected on the basis of their depth and breadth of experience, wisdom, integrity, ability to make independent analytical inquiries, understanding of the Company’s business environment, willingness to devote adequate time to board duties, the interplay of the candidate’s experience and skills with those of other directors and the extent to which the candidate would be a desirable addition to Board and any of its committees. Directors generally will not be nominated for re-election at any annual or special meeting held after their 75th birthday. In addition, the Company’s corporate governance guidelines require that directors limit their service on boards of directors of public companies to a total of four (including service on the Board). Other than the foregoing, there are no stated minimum criteria for director nominees, although the Nominating and Governance Committee may also consider such other factors as it may deem are in the best interests of the Company and its stockholders. The Nominating and Governance Committee does not have a policy regarding board diversity, but it takes diversity of professional experience and perspective within the pharmaceutical and biotechnology industries into account in identifying and selecting director nominees. In the case of incumbent directors whose terms of office are set to expire, the Nominating and Governance Committee reviews these directors’ overall service to the Company during their terms, including the number of meetings attended, level of participation, quality of performance and any other relationships and transactions that might impair the directors’ independence. In the case of new director candidates, the Nominating and Governance Committee also determines whether the nominee is independent for Nasdaq purposes, which determination is based upon applicable Nasdaq listing standards, applicable SEC rules and regulations and the advice of counsel, if necessary. The Nominating and Governance Committee then uses its network of contacts to compile a list of potential candidates, but may also engage, if it deems appropriate, a professional search firm. The Nominating and Governance Committee conducts any appropriate and necessary inquiries into the backgrounds and qualifications of possible candidates after considering the function and needs of the Board. The Nominating and Governance Committee meets to discuss and consider the candidates’ qualifications and then selects a nominee by majority vote which is typically recommended to the full board of directors.
The Nominating and Governance Committee will consider director candidates recommended by stockholders. The Nominating and Governance Committee does not intend to alter the manner in which it evaluates candidates, including the minimum criteria set forth above, based on whether or not the candidate was recommended by a stockholder. Stockholders who wish to recommend individuals for consideration by the Nominating and Governance Committee to become nominees for election to the Board may do so by delivering a written recommendation to the Nominating and Governance Committee at the following address: c/o Savara Inc., 900 S. Capital of Texas Highway, Las Cimas IV, Suite 150, Austin, Texas 78746, Attn: Secretary. Submissions must include the following information: the name, age, business address and residence address of the proposed nominee; a statement of the proposed nominee’s business experience and educational background; the proposed nominee’s principal occupation or employment; the class and number of shares of the Company’s capital stock beneficially owned by the proposed nominee; a detailed description of all relationships, arrangements or understandings between the proposing stockholder and the proposed nominee and any other person or persons (naming such person or persons) pursuant to which such proposed nomination is being made by the stockholder; a detailed description of all relationships, arrangements or understandings between the proposed nominee and any service-provider or supplier to, or competitor of, the Company’s; information regarding each of the criteria for board membership described above in sufficient detail to allow the Nominating and Governance Committee to evaluate the proposed nominee; and a statement from the proposed nominee that he or she is willing to be considered and willing to serve as a director if nominated and elected. The proposing stockholder must also include the following information with respect to such stockholder: documentation supporting that the proposing stockholder
90
Table of Contents
is a stockholder of the Company; the proposing stockholder’s name and address, as they appear on the Company’s books; and the class and number of shares of the Company’s capital stock beneficially owned by the proposing stockholder. If a stockholder submits a director recommendation in compliance with the procedure described above, the Nominating and Governance Committee will conduct an initial evaluation of the proposed nominee and, if it determines the proposed nominee may be a qualified candidate, the nominating and governance committee and one or more members of the management team will interview the proposed nominee to determine whether he or she might be suitable to be a director. If the Nominating and Governance Committee determines the proposed nominee would be a valuable addition to the Board, based on the criteria for board membership described above and the specific needs of the Board at the time, it will recommend to the Board such person’s nomination. In connection with its evaluation, the Nominating and Governance Committee may request additional information from the proposed nominee and/or the proposing stockholder. Separately, the Company’s bylaws contain provisions that address the process by which a stockholder may nominate an individual to stand for election to the Board at the Company’s annual meeting of stockholders. Such nominations may be made only if the stockholder has given timely written notice to the Company’s corporate secretary containing the information required by the Company’s bylaws. To be timely, such notice must be received at the Company’s principal executive offices not earlier than the 120th day, nor later than the close of business on the 90th day, prior to the first anniversary of the date of the preceding year’s annual meeting as first specified in the notice of meeting (without regard to any postponements or adjournments of such meeting after such notice was first sent), except that if no annual meeting was held in the previous year or the date of the annual meeting is more than 30 days earlier or later than such anniversary date, such notice must be received not earlier than the 120th day prior to the date of such annual meeting and not later than the close of business on the later of the 90th day prior to the date of such annual meeting or the 10th day following the date on which the Company first publicly announces the date of such meeting.
The Nominating and Governance Committee currently consists of Mr. Pikover, who serves as its chairman, Dr. McCracken and Mr. Elam. All members of the Nominating and Governance Committee are independent (as independence is currently defined in Nasdaq listing standards).
The board of directors of the Company may from time to time establish other committees.
2016 Savara Director Compensation
The table below shows all compensation earned by or paid to Savara’s non-employee directors during the year ended December 31, 2016.
| Name |
Fees Earned or Paid in Cash |
OptionAwards | Total | |||||||||
| Nevan Elam |
$ | — | $ | 11,993 | $ | 11,993 | ||||||
| Richard J. Hawkins |
$ | — | $ | 11,993 | $ | 11,993 | ||||||
| Yuri Pikover |
$ | — | $ | 11,993 | $ | 11,993 | ||||||
| Joseph S. McCracken |
$ | — | $ | 11,993 | $ | 11,993 | ||||||
91
Table of Contents
Compensation Committee Interlocks and Insider
Each member of the Compensation Committee is an “outside” director as that term is defined in Section 162(m) of the Internal Revenue Code, a “non-employee” director within the meaning of Rule 16b-3 of the rules promulgated under the Exchange Act and independent within the meaning of the independent director guidelines of Nasdaq. None of the Company’s executive officers serve as a member of the board of directors or compensation committee of any entity that has one or more executive officers who serves on the Company’s board of directors or Compensation Committee.
This section discusses the material components of the executive compensation program offered to Savara’s named executive officers identified below.
2016 Summary Compensation Table
The following table provides information regarding Savara’s named executive officers during the fiscal year ended December 31, 2016. These individuals are referred to elsewhere in this current report as the “named executive officers” of Savara.
| Name and Principal Position |
Year | Salary(1) | Option Awards(2) |
Non-Equity Incentive Plan Compensation |
All Other Compensation |
Total | ||||||||||||||||||
| Robert Neville |
2016 | $ | 302,500 | $ | 162,069 | $ | 20,000 | $ | 18,000 | $ | 502,569 | |||||||||||||
| Chief Executive Officer |
||||||||||||||||||||||||
| Taneli Jouhikainen |
2016 | $ | 302,500 | $ | 162,069 | $ | 32,000 | $ | 18,000 | $ | 514,569 | |||||||||||||
| President and Chief Operating Officer |
||||||||||||||||||||||||
| David Lowrance |
2016 | $ | 50,417 | $ | 118,405 | $ | — | $ | — | $ | 168,822 | |||||||||||||
| Chief Financial Officer |
||||||||||||||||||||||||
| (1) | Mr. Lowrance was hired on November 1, 2016. His annual salary as of December 31, 2016 was $302,500. |
| (2) | The amounts in the “Option Awards” column reflect the aggregate grant date fair value of stock options granted during the calendar year computed in accordance with the provisions of Accounting Standards Codification (ASC) 718, Compensation — Stock Compensation. The assumptions that Savara used to calculate these amounts are discussed in the notes to the unaudited interim condensed consolidated financial statements of Savara included elsewhere in this proxy statement/prospectus/information statement. These amounts do not reflect the actual economic value that will be realized by the named executive officer upon the vesting of the stock options, the exercise of the stock options, or the sale of the common stock underlying such stock options. |
Narrative Disclosure to Summary Compensation Table
The primary elements of compensation for Savara’s named executive officers are base salary, non-equity incentive plan awards and long-term, equity-based compensation awards. The named executive officers also participate in employee benefit plans and programs that Savara offers to its other full-time employees on the same basis.
Base Salary
The base salary payable to Savara’s named executive officers is intended to provide a fixed component of compensation that reflects the executive’s skill set, experience, role and responsibilities.
92
Table of Contents
Non-Equity Incentive Plan
Although Savara does not have a written bonus plan, the Savara Board sets performance targets annually for each of the named executive officers, and the named executive officers receive bonuses at the end of each year based on achievement of those targets.
Health, Welfare and Additional Benefits
Each of Savara’s named executive officers is eligible to participate in Savara’s employee benefit plans and programs, including medical, dental and vision benefits, flexible spending accounts with company contribution, long-term care benefits, and short- and long-term disability, 401k retirement plan with company match potential to the same extent as its other full-time employees, subject to the terms and eligibility requirements of those plans.
Although Savara does not have a formal policy with respect to the grant of equity incentive awards to its executive officers or any formal equity ownership guidelines applicable to them, Savara believes that equity grants provide its executives with a strong link to Savara’s long-term performance, create an ownership culture and help to align the interests of Savara’s executives and its stockholders. In addition, Savara believes that equity grants with a time-based vesting feature promote executive retention because this feature incentivizes executive officers to remain in Savara’s employment during the vesting period. In 2016, Savara granted Mr. Neville an option to purchase 250,000 shares of Savara common stock, Dr. Jouhikainen an option to purchase 250,000 shares of Savara common stock and Mr. Lowrance an option to purchase 217,710 shares of its common stock.
Grants of Plan-Based Awards
The following table presents the awards to Savara’s named executive officers in 2016.
| Name |
Grant Date |
Estimated Future Payouts Under Non- Equity Incentive Plan Awards |
All Other Option Awards: Number of Securities Underlying Options |
Exercise or Base Price of Option Awards |
Grant Date Fair Value |
|||||||||||||||||||||||
| Threshold | Target | Maximum | ||||||||||||||||||||||||||
| Robert Neville |
12/15/16 | $ | — | $ | 63,525 | $ | 90,750 | 250,000 | $ | 1.03 | $ | 162,069 | ||||||||||||||||
| Taneli Jouhikainen |
12/15/16 | $ | — | $ | 63,525 | $ | 90,750 | 250,000 | $ | 1.03 | $ | 162,069 | ||||||||||||||||
| David Lowrance |
10/25/16 | $ | — | $ | 63,525 | $ | 90,750 | 217,710 | $ | 0.88 | $ | 118,405 | ||||||||||||||||
2016 Outstanding Equity Awards at Year-End
The following table presents the outstanding equity awards held by Savara’s named executive officers as of December 31, 2016.
| Option Awards | Stock Awards | |||||||||||||||||||||||
| Name |
Number of Securities Underlying Unexercised Options Exercisable |
Number of Securities Underlying Unexercised Options Unexercisable |
Option Exercise price |
Option Expiration date |
Number of shares of stock that have not vested |
Market value of shares of stock that have not vested |
||||||||||||||||||
| Robert Neville |
|
— 75,000 55,259 170,000 |
|
|
250,000 225,000 55,258 0 |
|
$ $ $ $ |
1.03 0.85 0.38 0.38 |
|
|
12/15/2026 12/15/2025 12/16/2024 09/14/2022 |
|
27,845 | $ | 31,186 | |||||||||
| Taneli Jouhikainen |
|
— 75,000 90,000 |
|
|
250,000 225,000 0 |
|
$ $ $ |
1.03 0.85 0.38 |
|
|
12/16/2026 12/15/2025 12/14/2022 |
|
83,104 | $ | 93,076 | |||||||||
| David Lowrance |
— | 217,710 | $ | 0.88 | 11/1/2026 | |||||||||||||||||||
93
Table of Contents
Employment and Severance Agreements
Savara entered into executive employment agreements with each of Robert Neville, Taneli Jouhikainen, and David Lowrance on March 9, 2017, which supersede their existing employment agreements with Savara. The agreements are for an unspecified term and entitle each of Mr. Neville, Dr. Jouhikainen, and Mr. Lowrance to an annual base salary of $302,500. The agreements also provide that each executive will be eligible to receive an annual performance-based bonus of up to 30% of the executive’s base salary upon achievement of performance objectives to be determined by the Savara Board, compensation committee of the Savara Board, or their delegate. The amount of the performance-based bonus will be determined in the sole discretion of the Savara Board or the compensation committee of the Savara Board. Pursuant to the terms of the agreements, each of Mr. Neville, Dr. Jouhikainen, and Mr. Lowrance is subject to certain confidentiality obligations and is obligated to continue to comply with the agreement relating to proprietary information and inventions each executive has previously entered into with Savara. Further provisions of the agreements are discussed below in the section entitled, “Potential Payments Upon Termination of Employment or Change in Control.”
Potential Payments Upon Termination of Employment or Change in Control
Pursuant to the terms of their respective executive employment agreements, if Savara terminates Mr. Neville’s, Dr. Jouhikainen’s, or Mr. Lowrance’s employment with Savara other than for “cause” (as defined in the agreements), death, or disability, or Mr. Neville, Dr. Jouhikainen, or Mr. Lowrance resigns from such employment for “good reason” (as defined in the agreements) and such termination occurs outside of the period beginning three months prior to, and ending 12 months following, a “change of control” (as defined in the agreements) (the “change of control period”), then, subject to the executive timely signing and not revoking a separation agreement and release of claims agreement (and, for Mr. Lowrance, provided such termination occurs after August 1, 2017), each of Mr. Neville, Dr. Jouhikainen, and Mr. Lowrance would be entitled to receive:
| • | a lump sum payment equal to (i) six months of his then-current base salary plus (ii) a pro-rated portion of his target bonus based on the number of days he was employed by Savara during the relevant performance period; |
| • | reimbursements for payments the executive makes for continued healthcare coverage pursuant to COBRA until the earlier of (i) the date six months from the termination date or (ii) the date upon which he and/or his eligible dependents becomes covered under similar plans; and |
| • | accelerated vesting as to the number of shares that would have otherwise vested pursuant to his Company equity awards had he remained employed by Savara for 12 months following his termination date. |
The executive employment agreements also provide that if Savara terminates Mr. Neville’s, Dr. Jouhikainen’s, or Mr. Lowrance’s employment with Savara other than for cause, death, or disability, or Mr. Neville, Dr. Jouhikainen, or Mr. Lowrance resign from such employment for good reason and such termination occurs during the change of control period, then, subject to the executive timely signing and not revoking a separation agreement and release of claims agreement (and, for Mr. Lowrance, provided such termination occurs after August 1, 2017), each of Mr. Neville, Dr. Jouhikainen, and Mr. Lowrance would be entitled to receive:
| • | a lump sum payment equal to (i) 12 months of his then-current base salary plus (ii) 100% of his target bonus; |
| • | a taxable lump sum payment equal to the amount he would pay for continued healthcare coverage pursuant to COBRA for 12 months from the termination date; and |
| • | accelerated vesting as to 100% of his Company equity awards. |
94
Table of Contents
Notwithstanding the foregoing, pursuant to Mr. Lowrance’s executive employment agreement, if Savara terminates his employment with Savara other than for cause, death, or disability, or Mr. Lowrance resigns from such employment for good reason and such termination occurs on or prior to August 1, 2017, then, subject to Mr. Lowrance signing and not revoking a separation agreement and release of claims agreement, he would be entitled to receive the following, regardless of whether the termination occurs within or outside of the change of control period:
| • | a lump sum payment equal to (i) three months of his then-current base salary plus (ii) 25% of his target bonus; and |
| • | reimbursements for payments Mr. Lowrance makes for continued healthcare coverage pursuant to COBRA until the earlier of (i) the date three months from the termination date or (ii) the date upon which he and/or his eligible dependents becomes covered under similar plans. |
95
Table of Contents
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS OF SAVARA
Described below are transactions occurring since January 1, 2014 and any currently proposed transactions to which Savara was a party and in which
| • | The amounts involved exceeded or will exceed $120,000; and |
| • | A director, executive officer, holder of more than 5% of the outstanding capital stock of Savara, or any member of such person’s immediate family had or will have a direct or indirect material interest, other than compensation, termination and change of control arrangements that are described under the section titled “Executive Compensation” in this proxy statement/prospectus/information statement. |
Sales of Securities
2014 Convertible Debt Financing
From May 2014 to October 2014, Savara issued an aggregate principal amount of $10,000,000 in convertible promissory notes. These promissory notes accrued interest at a rate of 8% per annum. All of the convertible promissory notes issued in such financing were converted into shares of Series C preferred stock in December 2015 in connection with the Series C preferred stock financing described below. The aggregate principal amount of the convertible promissory notes and aggregate accrued interest of $1,006,246 converted into shares of Series C preferred stock at a 20% discount to the purchase price paid for the Series C preferred stock by other investors in the Series C preferred stock financing. The following table sets forth the names of Savara’s directors, executive officers and holders of more than 5% of Savara capital stock who participated in the convertible debt financing.
| Name |
Principal Amount |
|||
| Robert Neville |
$ | 25,000 | ||
| Entities affiliated with Yuri Pikover(1) |
$ | 1,000,000 | ||
| (1) | Convertible promissory note issued to 37Ventures, LLC. |
Series C Preferred Stock
In December 2015, Savara issued an aggregate of 1,423,482 shares of its Series C preferred stock at a purchase price of $5.2605 per share, and issued an additional 2,615,308 shares of its Series C preferred stock upon the conversion of convertible promissory notes with an aggregate principal and accrued interest of $11,006,246. In a subsequent closing in February 2016, Savara issued 413,792 shares of its Series C preferred stock at a purchase price of $5.2605 per share. Immediately prior to the effective time of the merger, each share of Savara Series A preferred stock and Series B preferred stock will be converted into one share of Savara common stock and each share of Savara Series C preferred stock will be converted into 1.01706 shares of Savara common stock. The following table sets forth the names of the Savara directors, executive officers and holders of more than 5% of Savara capital stock who participated in the Series C preferred stock financing.
| Name of Stockholder |
Shares of Series C Preferred Stock |
Aggregate Purchase Price |
||||||
| Robert Neville |
8,337 | $ | 37,127 | |||||
| Entities affiliated with Yuri Pikover(1) |
292,695 | $ | 1,261,781 | |||||
| (1) | Shares issued to 37Ventures, LLC. |
2016 Convertible Debt Financing
From July 2016 to August 2016, Savara issued an aggregate principal amount of $4,414,689 in convertible promissory notes. These promissory notes accrued interest at a rate of 8% per annum. The terms of these convertible promissory notes were amended in January 2017 to provide for the conversion of the notes into shares of Savara common stock immediately prior to, and conditioned upon, the closing of the merger with Mast. Upon conversion of these promissory notes, Savara will issue an aggregate of approximately 1.1 million shares of common stock.
96
Table of Contents
In connection with the sale and issuance of the convertible promissory notes, Savara issued stock purchase warrants exercisable for shares of Savara’s equity securities at a purchase price of $5.2605 per share upon the occurrence of a specified exercise event. Such exercise events include a change in control of Savara, an initial public offering of Savara stock, or a Regulation A offering of Savara stock. The terms of the warrants were amended in January 2017 to include the closing of the merger with Mast as an exercise event following which the warrants may be exercised. The number of shares of equity securities exercisable pursuant to the warrants is equal to 2.8515% of the principal amount of such holder’s convertible promissory note.
The following table sets forth the names of Savara’s directors, executive officers and holders of more than 5% of Savara’s capital stock who participated in the convertible debt financing.
| Name |
Principal Amount |
|||
| Joseph S. McCracken |
$ | 25,000 | ||
| Entities affiliated with Yuri Pikover(1) |
$ | 50,000 | ||
| (1) | Convertible promissory note issued to 37Ventures, LLC. |
Series B Preferred Stock Warrants
In connection with the sale and issuance of Savara’s Series B Preferred Stock in May 2012, Savara issued stock purchase warrants exercisable for shares of Savara’s equity securities at a purchase price of $3.12959 per share at any time prior the earliest to occur of (i) the close of business on May 30, 2017, (ii) a change in control of Savara, or (iii) 360 days following the closing of an initial public offering of Savara stock. Warrants to purchase an aggregate of 289,966 shares were outstanding as of December 31, 2016.
Stockholder Agreements
In December 2015, Savara entered into its Fourth Amended and Restated Investors’ Rights Agreement, or the Rights Agreement, and in July 2016, Savara entered into its Fifth Amended and Restated Right of First Refusal Agreement, or the ROFR Agreement, and its Third Amended and Restated Voting Agreement, or the Voting Agreement, with certain holders of its preferred stock and certain holders of its common stock. Such agreements provide for, among other things, voting rights and obligations, information rights, rights of first refusal and registration rights. The following directors, executive officers and holders of more than 5% of Savara capital stock and their affiliates are parties to these agreements:
| • | Robert Neville; |
| • | Taneli Jouhikainen; |
| • | Joseph S. McCracken; and |
| • | 37Ventures, LLC (Yuri Pikover, an affiliate of 37Ventures, LLC, is a member of Savara’s board of directors). |
The ROFR Agreement, the Voting Agreement and the Rights Agreement will terminate upon the closing of the merger with Mast.
Director and Executive Officer Compensation
For information regarding the compensation of Savara’s directors and executive officers, please see the section entitled “Management Following the Merger — Director Compensation” in this proxy statement/prospectus/information statement.
Policy for Approval of Related Person Transactions
While Savara does not have a formal written policy or procedure for the review, approval or ratification of related party transactions, Savara’s board of directors reviews and considers the interests of its directors, executive officers and principal stockholders in its review and consideration of transactions.
97
Table of Contents
INDEMNIFICATION OF DIRECTORS AND OFFICERS
Subsection (a) of Section 145 of the General Corporation Law of the State of Delaware, or the DGCL, empowers a corporation to indemnify any person who was or is a party or who is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the corporation) by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with such action, suit or proceeding if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe the person’s conduct was unlawful.
Subsection (b) of Section 145 empowers a corporation to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that the person acted in any of the capacities set forth above, against expenses (including attorneys’ fees) actually and reasonably incurred by the person in connection with the defense or settlement of such action or suit if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification shall be made in respect of any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or the court in which such action or suit was brought shall determine upon application that, despite the adjudication of liability but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
Section 145 further provides that to the extent a director or officer of a corporation has been successful on the merits or otherwise in the defense of any action, suit or proceeding referred to in subsections (a) and (b) of Section 145, or in defense of any claim, issue or matter therein, such person shall be indemnified against expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection therewith; that indemnification provided for by Section 145 shall not be deemed exclusive of any other rights to which the indemnified party may be entitled; and the indemnification provided for by Section 145 shall, unless otherwise provided when authorized or ratified, continue as to a person who has ceased to be a director, officer, employee or agent and shall inure to the benefit of such person’s heirs, executors and administrators. Section 145 also empowers the corporation to purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against any liability asserted against such person and incurred by such person in any such capacity, or arising out of his status as such, whether or not the corporation would have the power to indemnify such person against such liabilities under Section 145.
Section 102(b)(7) of the DGCL provides that a corporation’s certificate of incorporation may contain a provision eliminating or limiting the personal liability of a director to the corporation or its stockholders for monetary damages for breach of fiduciary duty as a director, provided that such provision shall not eliminate or limit the liability of a director (i) for any breach of the director’s duty of loyalty to the corporation or its stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (iii) under Section 174 of the DGCL or (iv) for any transaction from which the director derived an improper personal benefit.
98
Table of Contents
The Company’s amended and restated certificate of incorporation provides that to the fullest extent permitted by the Delaware General Corporation Law, (1) a director shall not be personally liable to the Company or its stockholders for monetary damages for breach of fiduciary duty as a director, and (2) the Company shall indemnify any director or officer made a party to an action or proceeding, whether criminal, civil, administrative or investigative, by reason of the fact of such person’s current or prior service as a director or officer of the Company, as a director or officer of any of the Company’s predecessors or any other enterprise per the Company or any of its predecessor’s request. The Company’s amended and restated bylaws provide that (a) the Company shall indemnify its directors and officers to the maximum extent and in the manner permitted by the Delaware General Corporation Law against expenses (including attorneys’ fees), judgments, fines, ERISA excise taxes, settlements and other amounts actually and reasonably incurred in connection with any proceeding, whether civil, criminal, administrative or investigative, arising by reason of the fact that such person is or was an agent of the corporation, subject to certain limited exceptions, (b) the Company shall advance expenses incurred by any director or officer prior to the final disposition of any proceeding to which the director or officer was or is or is threatened to be made a party promptly following a request therefore, subject to certain limited exceptions, and (c) the rights conferred in the Company’s amended and restated bylaws are not exclusive.
The Company entered into indemnification agreements with its directors and executive officers, in addition to the indemnification provided for in its amended and restated certificate of incorporation and bylaws, and intends to enter into indemnification agreements with any new directors and executive officers in the future.
The Company has purchased and intends to maintain insurance on behalf of any person who is or was a director or officer of the Company against any loss arising from any claim asserted against him or her and incurred by him or her in any such capacity, subject to certain exclusions. Pursuant to the terms of the Merger Agreement the Company purchased an insurance policy, which maintains in effect for six years from the closing the directors’ and officers’ liability insurance policies maintained by the Company prior to the Closing.
Pursuant to the terms of the Merger Agreement, the provisions relating to the indemnification and elimination of liability for monetary damages set forth in the certificate of incorporation and bylaws of the Company shall not be amended, repealed or otherwise modified for a period of six years’ time from the closing of the merger in a manner that would adversely affect the rights thereunder of individuals who, at or prior to the closing, were officers, directors, employees or agents of the Company.
99
Table of Contents
Reference is made to the financial statements and pro forma financial information relating to Savara contained in item 9.01 of this Current Report on form 8-K, which is incorporated herein by reference.
Item 9.01. Financial Statements and Exhibits.
| (a) | Financial Statements of Business Acquired. |
100
Table of Contents
| Savara |
||||
| Consolidated Financial Statements (Years Ended December 31, 2016, 2015 and 2014) |
||||
| Report of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm |
102 | |||
| 103 | ||||
| 104 | ||||
| Statements of Changes in Redeemable Convertible Preferred Stock and Stockholders’ Deficit |
105 | |||
| 106 | ||||
| 107 | ||||
| Serendex |
||||
| Consolidated Financial Statements |
||||
| 133 | ||||
| Consolidated Income Statement and Statement of Comprehensive Income |
134 | |||
| 135 | ||||
| 136 | ||||
| 137 | ||||
| 138 | ||||
| Unaudited Interim Consolidated Financial Statements |
||||
| Unaudited Consolidated Income Statement and Statement of Comprehensive Income |
155 | |||
| 156 | ||||
| 157 | ||||
| 158 | ||||
| 159 | ||||
101
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Savara Inc.,
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations and comprehensive loss, of changes in redeemable convertible preferred stock and stockholders’ deficit, and of cash flows present fairly, in all material respects, the financial position of Savara Inc. and its subsidiary as of December 31, 2016 and 2015, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2016 in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We conducted our audits of these financial statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
As discussed in Note 1 to the financial statements, the cost to develop and obtain regulatory approval for any drug is substantial and if adequate and timely financing alternatives are not available, the Company will need to re-evaluate its current operating plan.
/s/ PricewaterhouseCoopers LLP
Austin, Texas
March 10, 2017
102
Table of Contents
Consolidated Balance Sheets
(In thousands, except share amounts)
| As of December 31 | ||||||||
| 2016 | 2015 | |||||||
| Assets |
||||||||
| Current assets: |
||||||||
| Cash and cash equivalents |
$ | 13,373 | $ | 16,683 | ||||
| Grants and award receivable |
400 | — | ||||||
| Prepaid expenses and other current assets |
840 | 67 | ||||||
|
|
|
|
|
|||||
| Total current assets |
14,613 | 16,750 | ||||||
| Property and equipment, net |
793 | 1,104 | ||||||
| In-process R&D |
10,477 | — | ||||||
| Goodwill |
3,051 | — | ||||||
|
|
|
|
|
|||||
| Total assets |
$ | 28,934 | $ | 17,854 | ||||
|
|
|
|
|
|||||
| Liabilities, redeemable convertible preferred stock and stockholders’ deficit |
||||||||
| Current liabilities: |
||||||||
| Accounts payable |
$ | 536 | $ | 385 | ||||
| Accrued expenses and other liabilities |
2,477 | 430 | ||||||
| Current portion of capital lease obligation |
442 | 255 | ||||||
|
|
|
|
|
|||||
| Total current liabilities |
3,455 | 1,070 | ||||||
| Long-term liabilities: |
||||||||
| Accrued interest on convertible promissory notes |
151 | — | ||||||
| Convertible promissory notes |
3,448 | — | ||||||
| Put option derivative liability |
979 | — | ||||||
| Contingent consideration |
9,708 | — | ||||||
| Deferred tax liability |
2,305 | — | ||||||
| Capital lease obligation, net of current portion |
579 | 847 | ||||||
| Warrant liability |
303 | 274 | ||||||
| Other long-term liabilities |
20 | 6 | ||||||
|
|
|
|
|
|||||
| Total liabilities |
20,948 | 2,197 | ||||||
|
|
|
|
|
|||||
| Redeemable convertible preferred stock: |
||||||||
| Series A redeemable convertible preferred stock, $0.001 par value, 1,799,906 shares authorized, issued, and outstanding as of December 31, 2016 and 2015; $3,254 liquidation value as of December 31, 2016 |
3,232 | 3,230 | ||||||
| Series B redeemable convertible preferred stock, $0.001 par value, 6,000,000 shares authorized as of December 31, 2016 and 2015; 5,675,387 shares issued and outstanding as of December 31, 2016 and 2015; $17,762 liquidation value as of December 31, 2016 |
17,301 | 17,224 | ||||||
| Series C redeemable convertible preferred stock, $0.001 par value; 8,000,000 and 5,000,000 shares authorized as of December 31, 2016 and 2015, respectively; 4,452,582 and 4,038,790 shares issued and outstanding as of December 31, 2016 and 2015, respectively; $23,423 liquidation value as of December 31, 2016 |
23,328 | 22,531 | ||||||
|
|
|
|
|
|||||
| Total redeemable convertible preferred stock |
43,861 | 42,985 | ||||||
|
|
|
|
|
|||||
| Stockholders’ deficit: |
||||||||
| Common stock, $0.001 par value, 27,000,000 and 20,000,000 shares authorized as of December 31, 2016 and 2015, respectively; 5,396,883 and 2,041,552 shares issued and outstanding as December 31, 2016 and 2015, respectively |
5 | 2 | ||||||
| Additional paid-in capital |
3,117 | 153 | ||||||
| Accumulated other comprehensive loss |
(591 | ) | — | |||||
| Accumulated deficit |
(38,406 | ) | (27,483 | ) | ||||
|
|
|
|
|
|||||
| Total stockholders’ deficit |
(35,875 | ) | (27,328 | ) | ||||
|
|
|
|
|
|||||
| Total liabilities, redeemable convertible preferred stock, and stockholder’s deficit |
$ | 28,934 | $ | 17,854 | ||||
|
|
|
|
|
|||||
The accompanying notes are an integral part of these financial statements.
103
Table of Contents
Consolidated Statements of Operations and Comprehensive Loss
Years Ended December 31, 2016, 2015 and 2014
(In thousands, except share amounts)
| Year Ended December 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Grant and award revenue |
$ | 400 | $ | 54 | $ | 1,548 | ||||||
| Operating expenses: |
||||||||||||
| Research and development |
8,182 | 4,321 | 5,429 | |||||||||
| General and administrative |
2,820 | 1,650 | 1,560 | |||||||||
| Depreciation |
346 | 6 | 8 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total operating expenses |
11,348 | 5,977 | 6,997 | |||||||||
|
|
|
|
|
|
|
|||||||
| Loss from operations |
(10,948 | ) | (5,923 | ) | (5,449 | ) | ||||||
| Other income (expense): |
||||||||||||
| Investment income |
16 | 5 | 6 | |||||||||
| Interest expense |
(468 | ) | (2,590 | ) | (837 | ) | ||||||
| Loss on extinguishment of debt |
— | (226 | ) | — | ||||||||
| Foreign currency exchange gain/(loss) |
76 | — | — | |||||||||
| Change in fair value of financial instruments |
44 | (265 | ) | (2 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Total other expense |
(332 | ) | (3,076 | ) | (833 | ) | ||||||
| Loss before income taxes |
(11,280 | ) | (8,999 | ) | (6,282 | ) | ||||||
| Income tax benefit |
357 | — | — | |||||||||
|
|
|
|
|
|
|
|||||||
| Net loss |
$ | (10,923 | ) | $ | (8,999 | ) | $ | (6,282 | ) | |||
| Accretion of preferred stock classified as mezzanine equity |
(94 | ) | (183 | ) | (121 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss attributable to common stockholders |
(11,017 | ) | (9,182 | ) | (6,403 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Other comprehensive income: |
||||||||||||
| Gain (loss) on foreign currency translation |
(591 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Total Comprehensive Loss |
$ | (11,514 | ) | $ | (8,999 | ) | $ | (6,282 | ) | |||
|
|
|
|
|
|
|
|||||||
| Net loss per share: |
||||||||||||
| Basic and diluted |
$ | (3.29 | ) | $ | (5.55 | ) | $ | (4.26 | ) | |||
|
|
|
|
|
|
|
|||||||
| Weighted average common shares outstanding |
||||||||||||
| Basic and diluted |
3,348,647 | 1,653,259 | 1,503,058 | |||||||||
|
|
|
|
|
|
|
|||||||
The accompanying notes are an integral part of these financial statements.
104
Table of Contents
Consolidated Statements of Changes in Redeemable Convertible Preferred Stock and Stockholders’ Deficit
Years Ended December 31, 2016
(In thousands, except share amounts)
| Redeemable Convertible Preferred Stock | Stockholders’ Deficit | |||||||||||||||||||||||||||||||||||||||||||||||||||
| Redeemable Convertible Series A Preferred Stock |
Redeemable Convertible Series B Preferred Stock |
Redeemable Convertible Series C Preferred Stock |
Common Stock | Accumulated Other Comprehensive Loss |
||||||||||||||||||||||||||||||||||||||||||||||||
| Number of Shares |
Amount | Number of Shares |
Amount | Number of Shares |
Amount | Total | Number of Shares |
Amount | Additional Paid-In Capital |
Accumulated Deficit |
Total | |||||||||||||||||||||||||||||||||||||||||
| Balance on December 31, 2013 |
1,799,906 | $ | 3,206 | 5,675,387 | $ | 16,944 | — | $ | — | $ | 20,150 | 1,825,776 | $ | 2 | $ | 163 | $ | (12,202 | ) | — | $ | (12,037 | ) | |||||||||||||||||||||||||||||
| Issuance of restricted common stock |
— | — | — | — | — | — | — | 165,776 | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||
| Accretion of redeemable convertible preferred stock |
— | — | — | 121 | — | — | 121 | — | — | (121 | ) | — | — | (121 | ) | |||||||||||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | — | 141 | — | — | 141 | |||||||||||||||||||||||||||||||||||||||
| Net loss |
— | — | — | — | — | — | — | — | — | — | (6,282 | ) | — | (6,282 | ) | |||||||||||||||||||||||||||||||||||||
| Balance on December 31, 2014 |
1,799,906 | $ | 3,206 | 5,675,387 | $ | 17,065 | — | $ | — | $ | 20,271 | 1,991,552 | $ | 2 | $ | 183 | $ | (18,484 | ) | $ | — | $ | (18,299 | ) | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| Issuance of restricted common stock |
— | — | — | — | — | — | — | 50,000 | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||
| Accretion of redeemable convertible preferred stock |
— | 24 | — | 159 | — | — | 183 | — | — | (183 | ) | — | — | (183 | ) | |||||||||||||||||||||||||||||||||||||
| Issuance of redeemable convertible preferred stock |
— | — | — | — | 4,038,790 | 22,531 | 22,531 | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | — | 153 | — | — | 153 | |||||||||||||||||||||||||||||||||||||||
| Net loss incurred |
— | — | — | — | — | — | — | — | — | — | (8,999 | ) | — | (8,999 | ) | |||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| Balance on December 31, 2015 |
1,799,906 | $ | 3,230 | 5,675,387 | $ | 17,224 | 4,038,790 | $ | 22,531 | $ | 42,985 | 2,041,552 | $ | 2 | $ | 153 | $ | (27,483 | ) | $ | — | $ | (27,328 | ) | ||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| Accretion of redeemable convertible preferred stock |
— | 2 | — | 77 | — | 15 | 94 | — | — | (94 | ) | — | — | (94 | ) | |||||||||||||||||||||||||||||||||||||
| Exercise of options to acquire common stock |
— | — | — | — | — | — | — | 1,406 | — | 1 | — | — | 1 | |||||||||||||||||||||||||||||||||||||||
| Common stock issued for acquisition of Serendex assets and subsidiaries |
— | — | — | — | — | — | — | 3,353,925 | 3 | 2,848 | — | — | 2,851 | |||||||||||||||||||||||||||||||||||||||
| Issuance of Series C convertible preferred stock, net |
— | — | — | — | 413,792 | 782 | 782 | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||
| Stock-based compensation |
— | — | — | — | — | — | — | — | — | 209 | — | — | 209 | |||||||||||||||||||||||||||||||||||||||
| Foreign exchange translation adjustment |
— | — | — | — | — | — | — | — | — | — | — | (591 | ) | (591 | ) | |||||||||||||||||||||||||||||||||||||
| Net loss incurred |
— | — | — | — | — | — | — | — | — | — | (10,923 | ) | — | (10,923 | ) | |||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
| Balance on December 31, 2016 |
1,799,906 | $ | 3,232 | 5,675,387 | $ | 17,301 | 4,452,582 | $ | 23,328 | $ | 43,861 | 5,396,883 | $ | 5 | $ | 3,117 | $ | (38,406 | ) | $ | (591 | ) | $ | (35,875 | ) | |||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||||||||||||||
The accompanying notes are an integral part of these financial statements.
105
Table of Contents
Consolidated Statements of Cash Flows
Years Ended December 31, 2016, 2015 and 2014
(In thousands)
| Year Ended December 31, | ||||||||||||
| 2016 | 2015 | 2014 | ||||||||||
| Cash flows from operating activities: |
||||||||||||
| Net loss |
$ | (10,923 | ) | $ | (8,999 | ) | $ | (6,282 | ) | |||
| Adjustments to reconcile net loss to net cash used in operating activities: |
||||||||||||
| Depreciation |
346 | 6 | 8 | |||||||||
| Changes in fair value of financial instruments |
(44 | ) | 265 | 2 | ||||||||
| Loss on extinguishment of debt |
— | 226 | — | |||||||||
| Noncash interest |
199 | 642 | 405 | |||||||||
| Foreign currency gain/(loss) |
76 | — | — | |||||||||
| Accretion on discount to convertible promissory notes |
269 | 1,948 | 432 | |||||||||
| Stock-based compensation |
209 | 153 | 141 | |||||||||
| Changes in operating assets and liabilities: |
||||||||||||
| Tax refund receivable |
393 | — | — | |||||||||
| Grant and award receivable |
(400 | ) | 961 | 1,584 | ||||||||
| Prepaid expenses and other current assets |
(289 | ) | 214 | (111 | ) | |||||||
| Deferred rent |
14 | 6 | — | |||||||||
| Accounts payable and accrued expenses |
1,780 | (200 | ) | 309 | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in operating activities |
(8,370 | ) | (4,778 | ) | (3,512 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from investing activities: |
||||||||||||
| Purchase of property and equipment |
(8 | ) | — | (3 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Net cash used in investing activities |
(8 | ) | — | (3 | ) | |||||||
|
|
|
|
|
|
|
|||||||
| Cash flows from financing activity: |
||||||||||||
| Proceeds from issuance of convertible promissory notes |
4,415 | — | 10,000 | |||||||||
| Proceeds from issuance of Series C preferred stock, net |
782 | 8,773 | — | |||||||||
| Net proceeds from the exercise of common stock options |
1 | — | — | |||||||||
| Capital lease obligation principle payments |
(81 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Net cash provided by financing activities |
5,117 | 8,773 | 10,000 | |||||||||
|
|
|
|
|
|
|
|||||||
| Effect of exchange rate changes on cash and cash equivalents |
(49 | ) | — | — | ||||||||
| Increase in cash and cash equivalents |
(3,310 | ) | 3,995 | 6,485 | ||||||||
| Cash and cash equivalents beginning of period |
16,683 | 12,688 | 6,203 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash and cash equivalents end of period |
$ | 13,373 | $ | 16,683 | $ | 12,688 | ||||||
|
|
|
|
|
|
|
|||||||
| Non-cash financing and investing activities: |
||||||||||||
| Issuance of common stock for Serendex |
$ | 2,851 | $ | — | — | |||||||
| Net assets acquired in business combination of Serendex |
(12,375 | ) | — | — | ||||||||
| Contingent liability related to purchase of Serendex |
9,524 | — | — | |||||||||
| Accretion of Series A redeemable convertible preferred stock |
2 | 24 | — | |||||||||
| Accretion of Series B redeemable convertible preferred stock |
77 | 159 | 121 | |||||||||
| Accretion of Series C redeemable convertible preferred stock |
15 | — | — | |||||||||
| Conversion of convertible promissory notes into preferred stock |
— | 11,006 | — | |||||||||
| Extinguishment of put option |
— | 2,708 | — | |||||||||
| Equipment under capital lease |
— | (1,102 | ) | — | ||||||||
| Supplemental disclosure of cash flow information: |
||||||||||||
| Cash paid for income taxes |
$ | — | 2 | 21 | ||||||||
The accompanying notes are an integral part of these financial statements.
106
Table of Contents
Notes to Consolidated Financial Statements
December 31, 2016, 2015 and 2014
1. Description of Business and Basis of Presentation
Description of Business
Savara Inc. (“Savara,” the “Company,” or as used in the context of “we” or “us”) is a clinical stage specialty pharmaceutical company focusing on the development and commercialization of product candidates for patients with rare respiratory diseases, including cystic fibrosis (CF), and pulmonary alveolar proteinosis (PAP). Our lead clinical stage product candidate, AeroVanc, is an inhaled formulation of vancomycin, intended for the treatment of persistent methicillin-resistant Staphylococcus aureus (MRSA) lung infection in CF patients. Our second clinical stage product candidate, Molgradex, is an inhaled formulation of recombinant human granulocyte-macrophage colony-stimulating factor (GM-CSF), intended for the treatment of PAP. Savara was formed as a corporation in Delaware in 2007. The Company operates in one segment and has its principal offices in Austin, Texas.
On July 15, 2016, the Company completed the acquisition of certain assets, liabilities, and subsidiaries of Serendex A/S (“Serendex”), through its wholly owned subsidiary, Savara ApS, a limited liability company established under the laws in Denmark (see note 7). Serendex was a biopharmaceutical development company that advanced a pipeline and portfolio of novel inhalation therapies and related technologies for the treatment of severe pulmonary conditions. With this acquisition, Savara strengthened its pipeline of rare respiratory disease products.
Since inception, the Company has devoted substantially all of its efforts and resources to identifying and developing its product candidates, recruiting personnel, and raising capital. Savara has incurred operating losses and negative cash flow from operations and has no product revenue from inception to date. The Company has not yet commenced commercial operations.
Basis of Presentation
The consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States (“U.S. GAAP”) as defined by the Financial Accounting Standards Board (“FASB”).
Revision of Previous Financial Statements
We determined the amount of goodwill and in-process R&D reported were incorrectly transposed on the balance sheet at December 31, 2016 as included in Mast Therapeutics’ Registration Statement on Form S-4 filed on March 10, 2016. The balance of goodwill, originally reported as $10,477,000 should have been reported as $3,051,000 and the IPR&D, originally reported as $3,051,000 should have been reported as $10,477,000. We assessed the materiality of this error on our financial statements previously issued in accordance with United States Security and Exchange Commission (“SEC”) Staff Accounting Bulletin (“SAB”) No. 99, Materiality, codified in Accounting Standards Codification (“ASC”) 250, Presentation of Financial Statements, and concluded that it was not material to our previously issued financial statements. The error has been corrected on the balance sheet as of December 31, 2016 included in these financial statements.
2. Summary of Significant Accounting Policies
Liquidity
As of December 31, 2016, the Company had an accumulated deficit of approximately $38.4 million. The Company also had negative cash flow from operations of approximately $8.4 million during the year ended December 31, 2016. The cost to further develop and obtain regulatory approval for any drug is substantial and, as noted below, the Company may have to take certain steps to maintain a positive cash position. Accordingly, the Company will need additional capital to further fund development of, and seek regulatory approvals for, its product candidates and begin to commercialize any approved products.
The Company is currently focused primarily on the development of pulmonary drugs and believes such activities will result in the Company’s continued incurrence of significant research and development and other expenses related to those programs. If the clinical trials for any of the Company’s product candidates fail or produce unsuccessful results and those product candidates do not gain regulatory approval, or if any of the Company’s product candidates, if approved, fails to achieve market acceptance, the Company may never become profitable. Even if the Company achieves profitability in the future, it may not be able to sustain profitability in subsequent periods. The Company intends to cover its future operating expenses through cash and cash equivalents on hand and through a combination of equity offerings, debt financings, government or other third-party funding, and
107
Table of Contents
other collaborations and strategic alliances. The Company cannot be sure that additional financing will be available when needed or that, if available, financing will be obtained on terms favorable to the Company or its stockholders.
While the Company expects its existing cash and cash equivalents of $13.4 million as of December 31, 2016 will enable it to fund operations and capital expenditure requirements into 2018, the Company may have to delay, reduce, limit or terminate some or all of its development programs or future commercialization efforts or grant rights to develop and market product candidates that the Company might otherwise prefer to develop and market itself in order to maintain a positive cash position. Failure to obtain adequate financing could adversely affect the Company’s ability to operate as a going concern. If the Company raises additional funds from the issuance of equity securities, substantial dilution to existing stockholders may result. If the Company raises additional funds by incurring debt financing, the terms of the debt may involve significant cash payment obligations as well as covenants and specific financial ratios that may restrict the Company’s ability to operate its business.
The Company intends to raise additional capital through the issuance of additional equity and potentially through borrowings and strategic alliances with partner companies. However, if such financings are not available timely and at adequate levels, the Company will need to reevaluate its operating plans. Management is currently pursuing financing alternatives. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Principles of Consolidation
The consolidated financial statements of the Company are stated in U.S. dollars and are prepared using U.S. GAAP. These financial statements include accounts of the Company and its wholly owned subsidiary. The financial statements of the Company’s wholly owned subsidiary are recorded in its functional currency and translated into the reporting currency. The cumulative effect of changes in exchange rates between the foreign entity’s functional currency and the reporting currency is reported in Accumulated Other Comprehensive Income. All intercompany transactions and accounts have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires the Company to make certain estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Management’s estimates include those related to the accrual of research and development costs, the valuation of preferred and common shares, certain financial instruments recorded at fair value, stock-based compensation, and the valuation allowance for deferred tax assets. The Company bases its estimates on historical experience and on various other market-specific and relevant assumptions that it believes to be reasonable under the circumstances. Accordingly, actual results could be materially different from those estimates.
Risks and Uncertainties
The product candidates being developed by the Company require approvals from the U.S. Food and Drug Administration (“FDA”) or foreign regulatory agencies prior to commercial sales. There can be no assurance that the Company’s product candidates will receive the necessary approvals. If the Company is denied regulatory approval of its product candidates, or if approval is delayed, it may have a material adverse impact on the Company’s business, results of operations and its financial position.
The Company is subject to a number of risks similar to other life science companies, including, but not limited to, risks related to the successful discovery and development of drug candidates, raising additional capital, development of competing drugs and therapies, protection of proprietary technology and market acceptance of the Company’s products. As a result of these and other factors and the related uncertainties, there can be no assurance of the Company’s future success.
108
Table of Contents
Concentration of Credit Risk
Financial instruments, which potentially subject the Company to concentrations of credit risk, consist principally of cash and cash equivalents. The Company places its cash and cash equivalents with a limited number of high quality financial institutions and at times may exceed the amount of insurance provided on such deposits.
Cash and Cash Equivalents
Cash and cash equivalents consist of cash and institutional bank money market accounts with original maturities of three months or less when acquired and are stated at cost, which approximates fair value due to the highly liquid nature of these securities.
Grants and Award Receivables
Receivables arise from incurring allowable costs under federal grants or through the achievement of milestones under an award from a non-profit organization that have not been received as of the balance sheet dates. Since inception, the Company has never incurred losses on its grants and award receivables, and as such, the Company has no allowance for doubtful accounts as of December 31, 2016 as management deemed all outstanding grants and award receivable balances collectible.
Fair Value of Financial Instruments
The accounting standard for fair value measurements provides a framework for measuring fair value and requires disclosures regarding fair value measurements. Fair value is defined as the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date, based on the Company’s principal or, in absence of a principal, most advantageous market for the specific asset or liability.
The Company uses a three-tier fair value hierarchy to classify and disclose all assets and liabilities measured at fair value on a recurring basis, as well as assets and liabilities measured at fair value on a non-recurring basis, in periods subsequent to their initial measurement. The hierarchy requires the Company to use observable inputs when available, and to minimize the use of unobservable inputs, when determining fair value.
The three tiers are defined as follows:
| • | Level 1 – Observable inputs that reflect quoted market prices (unadjusted) for identical assets or liabilities in active markets; |
| • | Level 2 – Observable inputs other than quoted prices in active markets that are observable either directly or indirectly in the marketplace for identical or similar assets and liabilities; and |
| • | Level 3 – Unobservable inputs that are supported by little or no market data, which require the Company to develop its own assumptions. |
Financial instruments carried at fair value include cash and cash equivalents, certain warrants classified as liabilities, contingent consideration, and an embedded put option separated from the convertible promissory notes. These financial instruments are carried at fair value on a recurring basis.
The carrying amounts of accounts payable, accrued liabilities, and the convertible promissory notes host contract approximate fair value due to the highly liquid nature of these short-term instruments.
Net Loss per Share
Basic net loss per share is calculated by dividing the net loss by the weighted average number of shares of common stock outstanding during the period without consideration of common stock equivalents. Since the
109
Table of Contents
Company was in a loss position for all periods presented, diluted net loss per share is the same as basic net loss per share for all periods as the inclusion of all potential common shares outstanding would have been anti-dilutive.
Property and Equipment
Property and equipment are stated at cost, net of accumulated depreciation. Depreciation is determined on a straight-line basis over the estimated useful lives of the assets, which range from three to five years. Repairs and maintenance that do not improve or extend the useful life of the respective asset are charged to expense as incurred.
Equipment under Capital Lease
In 2015, the Company entered into a contract manufacturing arrangement that included the right to use specified equipment. Management concluded that the contract manufacturing arrangement contains an embedded lease of the specified equipment based on the facts and circumstances, including the Company’s ability to direct the use of the equipment and because management believes that it is remote that any party other than the Company will take more than a minor output produced by the equipment during the term of the arrangement. Management performed an analysis under ASC 840 to determine the proper accounting for the embedded lease and concluded that there is a capital lease because the present value of the minimum lease payments per the contract exceeds 90% of the fair value of the equipment. The capitalized equipment is depreciated on a straight-line basis over the lesser of the non-cancellable lease term or the useful life, and the lease obligation accrues interest at the incremental rate used in the present value analysis.
Debt Issuance Costs
Debt issuance costs are presented on the balance sheets as a direct deduction from the carrying amount of the debt liability. Debt issuance costs incurred in the years ended December 31, 2016 and 2015 were insignificant.
Patents and Intellectual Property
The Company currently expenses all patent application costs. As the Company’s products are currently under research and development and are not currently approved for market, costs incurred in connection with patent applications are expensed as incurred due to the uncertainty of the future economic benefits of the underlying patents and intellectual property.
Goodwill and Acquired In-Process Research and Development (IPR&D)
Goodwill and acquired IPR&D are not amortized but they are tested annually for impairment or more frequently if impairment indicators exist. The Company adopted accounting guidance related to annual and interim goodwill and acquired IPR&D impairment tests which allows the Company to first assess qualitative factors before performing a quantitative assessment of the fair value of a reporting unit. If determined to be necessary, the two-step impairment test shall be used to identify potential goodwill impairment and measure the amount of a goodwill impairment loss to be recognized (if any). The Company performed a qualitative assessment and concluded it was more likely than not that the fair value of the Company’s reporting unit is less than its carrying value, including goodwill. The $151,000 decrease in the carrying value of goodwill from the acquisition date, July 15, 2016, was due to foreign currency translation.
Tax Refund Receivable
Under Danish Tax Law, Denmark remits a research and development tax credit equal to 22% of qualified research and development expenditures, not to exceed established thresholds. The Company, through its acquisition of Serendex on July 15, 2016 (see note 7), acquired a receivable for a Danish tax credit earned by a subsidiary of Serendex in 2015 in the amount of $892,000. The amount was subsequently received in December 2016. As of December 31, 2016, the Company estimated an additional tax credit of $357,000 for research and
110
Table of Contents
development expenditures incurred during 2016 subsequent to the acquisition. As of December 31, 2016, the payment had not yet been received and a receivable of $357,000 was recorded on the balance sheet within prepaid expenses and other current assets.
Segment Reporting
Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision-making group, in making decisions on how to allocate resources and assess performance. Our chief operating decision maker is the chief executive officer. We have one operating segment, specialty pharmaceuticals within the respiratory system.
Redeemable Convertible Preferred Stock and Series B and C Warrants
The Series A, Series B, and Series C redeemable convertible preferred stock is classified in temporary equity as it is redeemable at the written request from the holders of at least two-thirds of the then outstanding shares of preferred stock, at any time after October 31, 2022. Additionally, certain outstanding warrants to purchase the Series B and Series C redeemable convertible preferred stock (“Series B Warrants” and the “Series C Warrants”) are classified as liabilities because the Series B and Series C redeemable convertible preferred stock are contingently redeemable.
Revenue Recognition
To date, the Company has recognized revenue solely from federal grants under the Small Business Innovation Research Program of the Department of Health and Human Services, National Institutes of Health (“NIH”, together the “Federal Grants”) and an award from the Cystic Fibrosis Foundation Therapeutics, Inc. (the “CFFT”), a non-profit organization (the “CFF Award”) as further described in Note 6. The Company has not generated any product revenue to date. The Company’s ability to generate product revenues, which the Company does not expect will occur for many years, if ever, will depend heavily on the successful development, regulatory approval and eventual commercialization of the Company’s product candidates. The Company records revenue related to the Federal Grants as qualifying costs are incurred, and when there is reasonable assurance that the conditions of the grant have been met and the grant will be received. The Company records revenue related to the CFF Award upon completion and achievement of defined milestones, and when there is reasonable assurance that the conditions of the award have been met and collectability is reasonably assured.
Accrued Research and Development Costs
Research and development costs are expensed as incurred. Research and development costs include, but are not limited to, salaries, benefits, travel, stock-based compensation, consulting costs, contract research service costs, laboratory supplies, contract manufacturing costs, and costs paid to other third parties that conduct research and development activities on the Company’s behalf.
The Company records the costs associated with clinical trials and manufacturing development as incurred. These costs are a significant component of the Company’s research and development expenses, as a substantial portion of the Company’s on-going research and development activities are conducted by third-party service providers, including contract research organizations (“CROs”).
The Company accrues for expenses resulting from obligations under contracts with CROs and consultants for which payment flows do not match the periods over which materials or services are provided to the Company. The Company’s objective is to reflect the appropriate expense in the financial statements by recognizing the expenses in the period in which services are performed and efforts are expended.
In the event advance payments are made to a CRO, the payments are recorded as a prepaid asset and amortized as services are performed and efforts are expended. As actual costs become known, the Company adjusts its
111
Table of Contents
accruals. Changes in these estimates that result in material changes to the Company’s accruals could materially affect the Company’s results of operations. There were no material changes to the estimates in the periods presented.
Stock-Based Compensation
The Company recognizes the cost of stock-based awards granted to employees based on the estimated grant-date fair values of the awards. The value of the portion of the award that is ultimately expected to vest is recognized as expense ratably over the requisite service period. The Company recognizes the compensation costs for awards that vest over several years on a straight-line basis over the vesting period (see Note 12). The Company recognizes the cost of stock-based awards granted to non-employees at their then-current fair values as services are performed, and such awards are remeasured through the counterparty performance date.
Manufacturing Commitments and Contingencies
The Company is subject to various manufacturing royalties and payments related to Molgradex. Upon the successful development, registration and attainment of approval by the proper health authorities, such as the FDA, in any territory except Latin America, Central America and Mexico, the Company must pay a royalty of three percent (3%) on annual net sales to the manufacturer of its Active Pharmaceutical Ingredients (“API”). Under this agreement with the API manufacturer, no signing fee or milestones are included in the royalty payments, and there is no minimum royalty. Additionally, Savara has a commitment to acquire a working cell bank and a master cell bank for approximately $2.0 million from this API manufacturer in the third quarter of 2017.
The Company is also subject to certain contingent milestone payments up to approximately 7.0 million euros based upon various development activities and regulatory approvals payable to the Company’s manufacturer of its nebulizer used to administer Molgradex. In addition to these milestones, the Company will owe a royalty to the manufacturer of its nebulizer based on net sales. The royalty rate ranges from three and a half percent (3.5%) to five percent (5%) depending on the device technology used by the Company to administer the product.
Income Taxes
The Company uses the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are recognized for the expected future tax consequences of temporary differences between the carrying amounts and the tax bases of assets and liabilities. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect of a change in tax rates on deferred tax assets and liabilities will be recognized in the period that includes the enactment date. A valuation allowance is established against the deferred tax assets to reduce their carrying value to an amount that is more likely than not to be realized.
Recent Accounting Pronouncements
In August 2014, the FASB issued Accounting Standards Update 2014-15, “Disclosure of Uncertainties about an Entity’s Ability to Continue as a Going Concern” (“ASU 2014-15”), which provides guidance on the presentation of management’s plans, when conditions or events raise substantial doubt about the entity’s ability to continue as a going concern within one year after the date that the financial statements are issued. ASU 2014-15 is effective for fiscal years ending after December 15, 2016. The adoption of this standard did not have a material impact on the Company’s financial statements.
In November 2015, the FASB issued Accounting Standards Update 2015-17, “Income Taxes, Balance Sheet Classification of Deferred Taxes” (“ASU 2015-17”), which eliminates the current requirement for reporting entities to present deferred tax liabilities and assets as current and noncurrent in a classified balance sheet. Instead, reporting entities will be required to classify all deferred tax assets and liabilities as noncurrent. This
112
Table of Contents
guidance is effective for fiscal years beginning after December 15, 2016. The Company has elected to early adopt this standard. The adoption of this standard did not have a material impact on the Company’s financial statements.
In February 2016, the FASB issued Accounting Standards Update 2016-02, “Leases” (“ASU 2016-02”). The update aims at making leasing activities more transparent and comparable, and requires substantially all leases be recognized by lessees on their balance sheet as a right-of-use asset and a corresponding lease liability, including leases currently accounted for as operating leases. The update also requires improved disclosures to help users of financial statements better understand the amount, timing and uncertainty of cash flows arising from leases. ASU 2016-02 is effective for fiscal years beginning after December 15, 2018 with early adoption permitted. The Company is currently evaluating the impact of the adoption of ASU 2016-02 on its financial statements.
In March 2016, the FASB issued Accounting Standards Update 2016-09, “Compensation - Stock Compensation: Improvements to Employee Share-Based Payment Accounting” (“ASU 2016-09”). ASU 2016-09 changes certain aspects of the accounting for share-based payment awards, including accounting and cash flow classification for excess tax benefits and deficiencies; income tax withholding obligations; forfeitures; and cash flow classification. ASU 2016-09 is effective for the Company for annual periods beginning after December 15, 2017, and interim periods within annual periods beginning after December 15, 2018 with early adoption permitted. The Company is currently evaluating the effect of this new guidance on its financial statements.
In March 2016, the FASB issued Accounting Standards Update 2016-6, “Derivatives and Hedging (Topic 815): Contingent Put and Call Options in Debt Instruments (“ASU 2016-06”) which simplifies the embedded derivative analysis for debt instruments containing contingent call or put options by removing the requirement to assess whether a contingent event is related to interest rates or credit risks. The guidance clarifies that a contingent put or call option embedded in a debt instrument would be evaluated for possible separate accounting as a derivative instrument without regard to the nature of the exercise contingency. The guidance is required to be applied on a modified retrospective basis to all existing and future debt instruments. This guidance is effective for fiscal years beginning after December 15, 2016, and interim periods within those fiscal years with early adoption permitted. The Company has elected to early adopt this standard. The adoption of this standard did not have a material impact on the Company’s financial statements.
In August 2016, the FASB issued Accounting Standards Update 2016-15, “Statement of Cash Flows (Topic 230): Classification of Certain Cash Receipts and Cash Payments” (“ASU 2016-15”), which intended to add or clarify guidance on the classification of certain cash receipts and payments on the statement of cash flows. The new guidance addresses cash flows related to the following: debt prepayment or extinguishment costs, settlement of zero coupon bonds, contingent consideration payments made after a business combination, proceeds from the settlement of insurance claims, proceeds from the settlement of corporate-owned life insurance policies and bank-owned life insurance policies, distributions received from equity method investees, beneficial interest in securitization transactions, and the application of predominance principle to separately identifiable cash flows. ASU 2016-15 is effective for the Company for annual periods beginning after December 15, 2018 with early adoption permitted. The Company is currently evaluating the effect of this new guidance on its financial statements.
In January 2017, the FASB issued Accounting Standards Update 2017-01, “Business Combinations (Topic 805): Clarifying the Definition of a Business” (“ASU 2017-01”), which intended to clarify the definition of a business with the objective of adding guidance to assist entities with evaluating whether transactions should be accounted for as acquisitions (or disposals) of assets or businesses. ASU 2017-01 is effective for the Company for annual periods beginning after December 15, 2018 with early adoption permitted. The Company is currently evaluating the effect of this new guidance on its financial statements.
In January 2017, the FASB issued Accounting Standards Update 2017-04, “Intangibles- Goodwill and Other (Topic 350): Simplifying the Test for Goodwill Impairment” (“ASU 2017-04”), which intended to eliminate Step 2 of the current two-step goodwill impairment test and requires only a one-step quantitative impairment test,
113
Table of Contents
whereby a goodwill impairment loss will be measured as the excess of a reporting unit’s carrying amount over its fair value (not to exceed the total goodwill allocated to the reporting unit). ASU 2017-04 is effective for the Company for annual or any interim goodwill impairment tests in fiscal years beginning after December 15, 2019. The Company has elected to early adopt this standard. The adoption of this standard did not have a material impact on the Company’s financial statements.
3. Property and Equipment, Net
Property and equipment, net consisted of (in thousands):
| 2016 | 2015 | |||||||
| Research and development equipment under capital lease |
$ | 1,102 | $ | 1,102 | ||||
| Equipment |
177 | 126 | ||||||
| Furniture and fixtures |
18 | 16 | ||||||
|
|
|
|
|
|||||
| Total property and equipment |
1,297 | 1,244 | ||||||
| Less accumulated depreciation |
(504 | ) | (140 | ) | ||||
|
|
|
|
|
|||||
| Property and equipment, net |
$ | 793 | $ | 1,104 | ||||
|
|
|
|
|
|||||
Depreciation expense for the years ended December 31, 2016, 2015 and 2014 was $346,000, $6,000 and $8,000, respectively and includes the amortization of the capital lease. The amount of accumulated amortization of capitalized leases as of December 31, 2016 was $339,000.
4. Convertible Promissory Notes
2014 Convertible Promissory Notes
During 2014, the Company borrowed $10,000,000 from several investors under convertible subordinate promissory notes (the “2014 Notes”). As described further below, on December 3, 2015, the 2014 Notes were converted into Series C redeemable convertible preferred stock (“Series C Preferred Stock”) in accordance with the Automatic Conversion provision of the 2014 Notes described below. See Note 10 for discussion of the issuance of Series C Preferred Stock.
The 2014 Notes had an 8.0% simple interest rate per annum computed on the basis of the actual number of days elapsed and a year of 365 days. All unpaid principal, together with any then accrued but unpaid interest was due and payable on the earliest of (i) December 31, 2015 (the “Maturity Date”), (ii) the closing of a change of control as defined, or (iii) the occurrence of an event of default, as defined (such earliest date is hereinafter referred to as “Maturity”). The 2014 Notes were pre-payable only with the written consent of the holders of a majority of the principal amount of the then-outstanding 2014 Notes. The following paragraphs describe the conversion features of the 2014 Notes, as they existed prior to the Automatic Conversion in 2015.
Automatic Conversion
The principal and any accrued interest automatically convert into shares of Qualified Financing Securities at the 2014 Note Conversion Price, upon the closing of a Qualified Financing (“Automatic Conversion”). In the event of an automatic conversion, the 2014 Notes are converted into that number of Qualified Financing Securities determined by dividing (i) the aggregate outstanding principal amount and accrued but unpaid interest by (ii) the 2014 Note Conversion Price. A Qualified Financing is defined as the next transaction (or series of related transactions) after the date of the 2014 Note and before Maturity in which the Company issues and sells shares of its preferred stock in exchange for aggregate gross proceeds of at least $5,000,000 (excluding amounts received upon conversion of indebtedness). Qualified Financing Securities means the equity securities issued by the Company in a Qualified Financing with such rights, preferences, privileges and restrictions, contractual or otherwise, as the securities issued by the Company in the Qualified Financing.
114
Table of Contents
The 2014 Note Conversion Price is the lesser of (a) (1) the lowest per share purchase price paid for (i) the Qualified Financing Securities by the investors in the Qualified Financing or (ii) the Next Round Securities issued to the investors in the Non-Qualified Financing in which the 2014 Note is converted times (2) 0.8 (i.e., a 20% discount); and (b) the quotient obtained by dividing (1) the Valuation Cap by (2) the Company’s fully diluted capitalization immediately prior to the initial closing of the Next Financing. For this purpose, the Valuation Cap is $50,000,000. Non-Qualified Financing means any transaction (or series of related transactions) after the date of this 2014 Note and before Maturity in which the Company issues and sells shares of its capital stock in any transaction that is not deemed to be a Qualified Financing. Next Round Securities means the equity shares sold in a Non-Qualified Financing.
Voluntary Conversion
In the event that the Company consummates a Non-Qualified Financing, at the option of each holder or holders of a majority of the outstanding aggregate principal amount, all or part of the outstanding principal and any accrued interest may be converted into Next Round Securities. A Non-Qualified Financing is any transaction (or series of related transactions) after the date of this 2014 Note and before Maturity in which the Company issues and sells shares of its capital stock in any transaction that is not deemed to be a Qualified Financing at the applicable 2014 Note Conversion Price as defined above.
Change in Control Conversion
In the event of a Change of Control after the date of the 2014 Note but prior to Maturity, at the option of each holder or holders of a majority of the outstanding aggregate principal amount, all or part of the outstanding principal amount and any accrued interest, (i) may be converted into the number of shares of Series B redeemable convertible preferred stock (“Series B Preferred Stock”) determined by dividing (x) the aggregate outstanding principal amount and any accrued interest by (y) the quotient obtained by dividing (1) the Valuation Cap by (2) the Company’s capital stock outstanding immediately prior to such Change of Control. Change of Control means any liquidation, dissolution or winding up of the Company, either voluntary or involuntary, and shall be deemed to be occasioned by, or to include, (i) a merger or consolidation of the Company into or with another entity after which the stockholders of the Company immediately prior to such transaction do not own, immediately following the consummation of the transaction by virtue of their shares in the Company or securities received in exchange for such shares in connection with the transaction, a majority of the voting power of the surviving entity in proportions substantially identical to those that existed immediately prior to such transaction and with substantially the same rights, preferences, privileges and restrictions as the shares they held immediately prior to the transaction, (ii) the sale, transfer or other disposition (but not including a transfer or disposition by pledge or mortgage to a bona fide lender) of all or substantially all of the assets of the Company (other than to a wholly owned subsidiary), or (iii) the sale or transfer by the Company or its stockholders of more than 50% of the voting power of the Company in a transaction or series of related transactions other than in a transaction or series of transactions effected by the Company primarily for financing purposes.
IPO Conversion
The entire outstanding principal amount plus any accrued interest automatically converts into shares of common stock of the Company at the IPO Conversion Price. The IPO Conversion Price is the quotient obtained by dividing (1) the Valuation Cap ($50,000,000) by (2) the Company’s fully diluted capitalization immediately prior to the consummation of the Initial Public Offering.
Maturity Date Conversion
The entire outstanding principal amount and any accrued interest automatically converts into shares of Series B Preferred Stock at the Series B Price upon the close of business of the Maturity Date. In the event of an automatic conversion pursuant to this provision, this 2014 Note converts into that number of Series B Preferred Stock determined by dividing (i) the aggregate outstanding principal amount of this 2014 Note plus any accrued interest by (ii) the Series B Price. The Series B Price is approximately $3.13 as adjusted for stock dividends, stock splits, recapitalizations and other similar events.
115
Table of Contents
Accounting for 2014 Notes
Management determined that the automatic conversion upon a Qualified Financing or Non-Qualified Financing as defined above represents, in substance, a put option (redemption feature) designed to provide the investor with a fixed monetary amount, settleable in shares. Management determined that this put option should be separated and accounted for as a derivative, primarily because the put option was issued at a substantial discount and meets the net settlement criterion.
The put option, with a fair value of $2,562,000 at inception, was initially recorded as a derivative liability on the balance sheet and a corresponding discount to the 2014 Notes. The discount was accreted to interest expense on the statements of operations and comprehensive loss over the term of the 2014 Notes using the effective interest rate method. The Company recorded interest expense of $0, $1,948,000 and $432,000 during 2016, 2015 and 2014 through the date of the Automatic Conversion, respectively, related to the accretion of the discount. The derivative liability was recorded at fair value as of December 31, 2014, and immediately prior to the Automatic Conversion with changes in fair value recognized in the statements of operations and comprehensive loss.
Automatic Conversion into Series C Preferred Stock
On December 3, 2015, the date of the Automatic Conversion, the 2014 Notes and separated put option liability were surrendered in exchange for Series C Preferred Stock. The debt host contract and separated derivative liability were both subject to extinguishment accounting, and a loss in the amount of $226,000 was recorded in the Statement of Operations and Comprehensive Loss for the year ended December 31, 2015. The loss was calculated as the difference between the net book value of the 2014 Notes plus the fair value of the put option immediately prior to the Automatic Conversion, and the fair value of the Series C Preferred Stock into which the 2014 Notes were converted.
2016 Convertible Promissory Notes
During 2016, the Company borrowed approximately $4.4 million from several investors under convertible subordinate promissory notes (the “2016 Notes”). The 2016 Notes accrues interest at 8.0% per annum computed on the basis of the actual number of days elapsed and a 365-day year. All unpaid principal, together with any then accrued but unpaid interest is due and payable on the earliest of (i) June 30, 2018 (the “Maturity Date”), (ii) the closing of a change of control as defined, or (iii) the occurrence of an event of default, as defined (such earliest date is hereinafter referred to as Maturity). The 2016 Notes are prepayable only with the written consent of the holders of a majority of the principal amount of the then-outstanding 2016 Notes. Of the total convertible notes, $1.5 million is due to a related party, Sorana A/S, the majority owner of Serendex, which holds approximately 15.2% of the Company’s fully diluted common stock pursuant to the Business Transfer Agreement effective on July 15, 2016 (see note 7). The following paragraphs describe the conversion features of the 2016 Notes.
Automatic Conversion
The principal and any accrued interest automatically convert into shares of Qualified Private Placement Financing Securities at the 2016 Note Conversion Price, upon the closing of a Qualified Private Placement Financing (“Private Placement Automatic Conversion”). In the event of a Private Placement Automatic Conversion, the 2016 Notes are converted into a number of Qualified Private Placement Financing Securities determined by dividing (i) the aggregate outstanding principal amount and accrued but unpaid interest by (ii) the 2016 Note Conversion Price. A Qualified Private Placement Financing is defined as the next Private Placement transaction (or series of related transactions) after the date of the 2016 Note and before Maturity in which the Company issues and sells shares of its preferred stock in exchange for aggregate gross proceeds of at least $5,000,000 (excluding amounts received upon conversion of indebtedness). Private Placement means any equity
116
Table of Contents
financing transaction (or series of related transactions) pursuant to a private placement exempt from the registration requirements of the Securities Act, other than pursuant to the exemption provided by Regulation A under the Securities Act (i.e., not a Regulation A Offering or the Initial Public Offering).
The Note Conversion Price is the lesser of (A) (i) the price per share of the Next Round Securities, Qualified Financing Shares or Regulation A Offering Shares, as the case may be, times (ii) 0.8 (i.e. a 20% discount), or (B) the quotient obtained by dividing $125,0000,000 (the “Valuation Cap”) by the Company’s fully diluted capitalization immediately prior to the initial closing of the Qualified Financing, Non-Qualified Financing, Qualified Regulation A Offering or Non-Qualified Regulation A Offering in which the Notes are converted. Non-Qualified Private Placement Financing means any transaction (or series of related transactions) after the date of the 2016 Note and before Maturity in which the Company issues and sells shares of its capital stock in any Private Placement transaction that is not deemed to be a Qualified Private Placement Financing. Next Round Securities means the equity shares sold in a Non-Qualified Private Placement Financing.
The entire outstanding principal amount of the 2016 Notes and any accrued but unpaid interest will be converted automatically into shares of Regulation A Securities at the Note Conversion Price upon the closing of a Qualified Regulation A Offering. In the event of an automatic conversion under a Qualified Regulation A Offering, the 2016 Notes will be converted into that number of Regulation A Securities determined by dividing (i) the aggregate outstanding principal amount of the 2016 Note and any accrued but unpaid interest by (ii) the Note Conversion Price. A Qualified Regulation A Offering means a Regulation A Offering with gross proceeds to the Company of at least $5,000,000 in one or more closings during a twelve-month period, excluding amounts received on conversion of the 2016 Notes.
Voluntary Conversion
In the event that the Company consummates a Non-Qualified Private Placement Financing, at the option of each holder or holders of a majority of the outstanding aggregate principal amount, all or part of the outstanding principal and any accrued interest may be converted into Next Round Securities. A Non-Qualified Private Placement Financing is any transaction (or series of related transactions) after the date of the 2016 Notes and before Maturity in which the Company issues and sells shares of its capital stock in any Private Placement transaction that is not deemed to be a Qualified Private Placement Financing at the applicable 2016 Note Conversion Price as defined above.
In the event that the Company consummates a Non-Qualified Regulation A Offering (i) at the option of the Holder, but subject to the consent of the Board, all or part of the outstanding principal amount of the 2016 Notes and any accrued but unpaid interest may be converted into Regulation A Securities, and (ii) at the option of the Majority Holders, all or part of the outstanding principal amount of the 2016 Notes and any accrued but unpaid interest will be converted into shares of Regulation A Securities. In the event of such conversion, the 2016 Notes will be converted into that number of shares of Regulation A Securities determined by dividing (x) the aggregate outstanding principal amount of the 2016 Notes and any accrued but unpaid interest by (y) the Note Conversion Price. A Non-Qualified Regulation A Offering means the closing of a Regulation A Offering with gross proceeds to the Company of less than $5,000,000, excluding amounts received on conversion of the 2016 Notes.
Change in Control Conversion
In the event of a Change of Control after the date of the 2016 Notes but prior to Maturity, at the option of each holder or holders of a majority of the outstanding aggregate principal amount, all or part of the outstanding principal amount and any accrued interest, (i) may be converted into the number of shares of Series C Redeemable Convertible Preferred Stock (“Series C Preferred Stock”) determined by dividing (x) the aggregate outstanding principal amount and any accrued interest by (y) the quotient obtained by dividing (1) the Valuation Cap ($125,000,000) by (2) the Company’s capital stock outstanding immediately prior to such Change of Control.
117
Table of Contents
A Change of Control means any liquidation, dissolution or winding up of the Company, either voluntary or involuntary, and shall be deemed to be occasioned by, or to include, (i) a merger or consolidation of the Company into or with another entity after which the stockholders of the Company immediately prior to such transaction do not own, immediately following the consummation of the transaction by virtue of their shares in the Company or securities received in exchange for such shares in connection with the transaction, a majority of the voting power of the surviving entity in proportions substantially identical to those that existed immediately prior to such transaction and with substantially the same rights, preferences, privileges and restrictions as the shares they held immediately prior to the transaction, (ii) the sale, transfer or other disposition (but not including a transfer or disposition by pledge or mortgage to a bona fide lender) of all or substantially all of the assets of the Company (other than to a wholly-owned subsidiary), or (iii) the sale or transfer by the Company or its stockholders of more than 50% of the voting power of the Company in a transaction or series of related transactions other than in a transaction or series of transactions effected by the Company primarily for financing purposes.
IPO Conversion
Upon an Initial Public Offering of the Company’s common stock, the entire outstanding principal amount plus any accrued interest under the 2016 Notes automatically converts into shares of Company common stock at the IPO Conversion Price. The IPO Conversion Price means the lesser of the (x) quotient obtained by dividing (1) the Valuation Cap ($125,000,000) by (2) the Company’s fully diluted capitalization immediately prior to the consummation of the Initial Public Offering or (y) quotient obtained by dividing (1) the pre-money valuation of the Company approved by the Board of Directors in connection with the Initial Public Offering, by (2) the Company’s fully diluted capitalization immediately prior to the consummation of the Initial Public Offering.
Maturity Date Conversion
The entire outstanding principal amount and any accrued interest under the 2016 Notes automatically converts into shares of Series C Preferred Stock at the Series C Price upon the close of business of the Maturity Date. In the event of such automatic conversion, the 2016 Notes convert into that number of Series C Preferred Stock determined by dividing (i) the aggregate outstanding principal amount of the 2016 Notes plus any accrued interest by (ii) the Series C Price. The Series C Price is $5.26 as adjusted for stock dividends, stock splits, recapitalizations and other similar events.
Accounting for 2016 Notes
Management determined that the automatic conversion upon a Qualified Private Placement Financing, a Qualified Regulation A Offering, a Non-Qualified Private Placement Financing, or a Non-Qualified Regulation A Offering as defined above represents, in substance, a put option (redemption feature) designed to provide the investor with a fixed monetary amount, settleable in shares. Management determined that this put option should be separated and accounted for as a derivative primarily because the put option was issued at a substantial discount and meets the net settlement criterion.
The put option, with a fair value of $977,000 at inception, was initially recorded as a derivative liability on the accompanying balance sheet and a corresponding discount to the 2016 Notes. The Company is accreting the discount to interest expense on the statement of operations and comprehensive loss over the term of the 2016 Notes using the effective interest rate method. The Company recorded interest expense of $269,000 during the year ended December 31, 2016 related to the accretion of the discount attributed to the put option liability and Series C Warrants (see note 9). The derivative liability was recorded at fair value at issuance of the 2016 Notes with changes in fair value recognized in the statement of operations and comprehensive loss. The change in fair value from the date of issuance through December 31, 2016 was immaterial.
118
Table of Contents
5. Litigation
The Company is not currently a party to any litigation, nor is the Company aware of any pending or threatened litigation that management believes would materially affect the Company’s business, operating results, financial condition or cash flows. The Company’s industry is characterized by frequent claims and litigation, including claims regarding patent and other intellectual property rights, as well as for product liability. As a result, in the future, the Company may be involved in various legal proceedings from time to time.
6. Federal Grants and CFF Award
On March 27, 2013, the Company was issued an additional Federal Grant from the NIH, grant number 2R44HL112393-02, “Development of Inhaled Vancomycin for Treatment of MRSA Infections in CF” in the amount of $3,986,000 with a project period from March 1, 2013 through February 29, 2016. The Company has incurred expenses and recognized associated revenue of $0, $54,000 and $548,000 related to the Federal Grants for the years ended December 31, 2016, 2015 and 2014, respectively. As of December 31, 2016 and 2015, there were no amounts related to the Federal Grants included in grants and award receivable in the accompanying balance sheets. All amounts recognized as revenue under the Federal Grants through December 31, 2016 have been collected.
In September 2013, the Company received a $1.7 million CFF Award from the CFFT. The CFF Award includes disbursements to the Company based on the achievement of certain milestones. For the years ended December 31, 2016, 2015 and 2014, the Company recognized $400,000, $0 and $1,000,000 in revenue related to the CFF award, respectively. The Company is subject to certain royalty payments due to the CFFT under the CFF Award based on commercialization of the Company’s product and either the achievement of certain sales volumes or a Change in Control Transaction, as defined below. As of December 31, 2016 and 2015, $400,000 and $0, related to the CFF Award were included in grants and award receivable in the accompanying balance sheets. The $400,000 award receivable recorded at December 31, 2016 was subsequently collected in January 2017.
Commercial Approval Royalty
A royalty is payable to the CFFT equal to three (3) times the amount of the CFF Award upon approval of the Company’s product for commercial use. The royalty is payable in equal installments of 33% due 60 days after first commercial sale; 33% due 90 days of the first anniversary of the first commercial sale; and 34% due within 90 days of 2nd anniversary of first commercial sale. This royalty will be reduced upon Change in Control Transaction payments as described below. As the Company’s product has not yet been approved for commercial use, the Company has not recorded a liability for the commercial approval royalty.
Additional Royalties
In addition, if net sales exceed $50.0 million for any calendar year occurring during the first five years after the first commercial sale, the Company must remit payment to the CFFT equal to one (1) times the CFF Award. Furthermore, if net sales exceed $100.0 million for any calendar year occurring during the first five years after first commercial sale, the Company must remit an additional payment to the CFFT equal to one (1) times the CFF Award. Given the Company has not recognized any sales from the Company’s product, the Company has not recorded a liability for any amounts due as additional royalties.
Change in Control Royalty
Upon a Change in Control Transaction, as defined below, occurring prior to the second anniversary date of the effective date of the CFF Award, September 30, 2015, the Company must remit a royalty payment to the CFFT equal to 5% of the proceeds from the Change in Control Transaction but not to exceed an amount equal to two times the CFF Award proceeds received. Upon a Change in Control Transaction occurring after the second anniversary date of the effective date of the CFF Award, the Company must remit a royalty payment to the CFFT equal to 5% of the proceeds from the Change in Control Transaction but not to exceed an amount equal to three times the CFF Award.
119
Table of Contents
A Change in Control Transaction is defined as the consummation (single or series of transactions) constituting (i) merger, share exchange, or other reorganization; (ii) sale by one of more stockholders of a majority voting power in the Company; or (iii) sale of substantially all of the assets of the Company. The Company has determined that a change of control is not probable and as such, has not recorded a liability for the change in control royalty.
The CFF Award may not be assigned by any party (other than to an affiliate or to a successor to substantially all of such party’s assets or business to which the CFF Award relates) without the consent of the other party.
If the Company initiates an “Interruption,” as defined under the CFF Award, for more than one year at any time before the first commercial sale of the product under the AeroVanc Program, the Company ceases to conduct, or has ceased to use commercially reasonable efforts to advance the research and development or commercialization of the AeroVanc Program, the Company shall transfer an exclusive, worldwide license to the CFFT of the Company’s research and development of the product under the AeroVanc Program limited to the right to manufacture, have manufactured, license, sell, use, support, offer to sell, any related invention from the Company’s AeroVanc Program.
7. Acquisition of Serendex Pharmaceuticals
On May 13, 2016, the Company entered into a Business Transfer Agreement with Serendex under which Serendex agreed to sell, transfer and assign to Savara all of its assets and subsidiaries, certain of its contracts, and certain of its employees and liabilities. Serendex was a limited liability company incorporated in Denmark and was listed on the Oslo Stock Exchange until May 4, 2016. On July 15, 2016, the Company completed the acquisition of Serendex through its wholly-owned subsidiary, Savara ApS, a limited liability company established under the laws in Denmark.
The Serendex Acquisition was an important step in fulfilling Savara’s vision to become a specialty pharmaceutical company focused on rare respiratory diseases. Serendex was a biopharmaceutical development company that was advancing a pipeline and portfolio of novel inhalation therapies for the treatment of severe pulmonary conditions. Through this acquisition, Savara gained access to the late-stage Molgradex for the treatment of PAP, with a Phase 2/3 clinical study ongoing in the EU and Japan. In addition to Molgradex, Savara gained access to an experienced development team familiar with all aspects of the Molgradex program.
For the purchase consideration, Savara agreed to provide the seller with 3,353,925 shares of Savara’s common stock. In addition to the purchase consideration shares, Savara Inc. agreed to pay the seller (i) $5,000,000 upon receipt of marketing approval of the medicinal product Molgradex, an inhalation formulation of recombinant human GM-CSF for the treatment of pulmonary alveolar proteinosis (the Product) by the European Medicines Agency, (ii) $15,000,000 upon receipt of marketing approval of the Product by the United States Food and Drug Administration, and (iii) $1,500,000 upon receipt of marketing approval of the Product by the Japanese Pharmaceuticals and Medical Devices Agency (the Contingent Milestone Payments). The Company estimated the likelihood of approval in each region, separately, based on the product candidate’s current phase of development and utilizing published studies of clinical development success rates for comparable non-oncology orphan drugs. The present value of the potential cash outflows from the probability weighted Contingent Milestone Payments was then estimated by taking into consideration that the Contingent Milestone Payments are similar to a business expense of the Company and would be senior to any Company debt obligations. The resulting weighted average present value factor was applied to discount the probability adjusted Contingent Milestone Payments for each region to derive the fair value of the Contingent Milestone Payments.
The Company accounted for the acquisition as a business combination by applying the acquisition method, which requires the assets acquired and liabilities assumed be recorded at fair value with limited exceptions. The Company used the Multi-Period Excess Earnings Model (MPEEM), a form of the income approach to value the in-process research and development intangible asset. The excess of the purchase price over the assets acquired and liabilities assumed represents goodwill. The goodwill is primarily attributable to the synergies expected to arise after the acquisition and is not expected to be deductible for tax purposes. The following table summarizes
120
Table of Contents
the consideration that the Company paid for Serendex and the amounts of the assets acquired and liabilities assumed recognized at the acquisition date:
| Purchase Consideration | (In thousands) | |||
| Fair value of Savara common stock issued for the acquisition |
$ | 2,851 | ||
| Estimated fair value of Contingent Milestone Payments |
9,524 | |||
|
|
|
|||
| Fair value of total consideration |
$ | 12,375 | ||
|
|
|
|||
| Assets acquired and liabilities assumed |
||||
| Inventory |
$ | 18 | ||
| Income tax receivable |
872 | |||
| Property and equipment, net |
28 | |||
| Current liabilities |
(320 | ) | ||
| Deferred tax liability |
(2,419 | ) | ||
| In-process research and development intangible asset |
10,994 | |||
|
|
|
|||
| Total assets acquired and liabilities assumed |
9,173 | |||
| Goodwill |
3,202 | |||
|
|
|
|||
| $ | 12,375 | |||
|
|
|
|||
Pro Forma Financial Information
The following pro forma financial information reflects the consolidated results of operations of the Company for the year ended December 31, 2016 as if the acquisition of Serendex had taken place on January 1, 2015 (in thousands). The pro forma financial information is not necessarily indicative of the results of operations as they would have been had the transactions been effected on the assumed date. Included in the Savara consolidated statement of operations for the year ended December 31, 2016 is $0 of revenue and $2,778,000 of net loss before income tax generated by the Serendex business since July 15, 2016, the acquisition date.
| Year Ended December 31, 2016 |
Year Ended December 31, 2015 |
|||||||
| Net revenues |
$ | 400 | $ | 54 | ||||
| Net loss |
$ | 17,748 | $ | 18,072 | ||||
8. Commitments and Contingencies
As of December 31, 2016, the Company leased its office facilities under a non-cancellable operating lease. The lease term was extended for a period of 48 months, commencing on December 1, 2015, and expiring on November 30, 2019. The Company recognizes rent expense on a straight-line basis over the operating lease term. The lease is cancellable three years after execution of the lease if the Company notifies the property owner of its intention to cancel the lease by the end of second year of the lease. The future minimum annual lease payments under the operating lease are as follows (in thousands):
| Year ending December 31, |
||||
| 2017 |
$ | 172 | ||
| 2018 |
174 | |||
| 2019 |
161 | |||
|
|
|
|||
| Total minimum lease payments |
$ | 507 | ||
|
|
|
121
Table of Contents
Rent expense for the years ended December 31, 2016, 2015 and 2014 was $132,000, $106,000 and $96,000, respectively.
As of December 31, 2016, the Company leases certain research and development equipment as part of a contract manufacturing arrangement. The leased equipment is accounted for as a capital lease, and the present value of the future minimum lease payments are recorded as a liability on the balance sheet as of December 31, 2016. The future minimum annual lease payments under the capital lease are as follows (in thousands):
| Year ending December 31, |
||||
| 2017 |
$ | 486 | ||
| 2018 |
312 | |||
| 2019 |
313 | |||
|
|
|
|||
| Total minimum lease payments |
1,111 | |||
|
|
|
|||
| Less: imputed interest |
(90 | ) | ||
|
|
|
|||
| Total capital lease obligation |
$ | 1,021 | ||
|
|
|
The Company is also subject to certain contingent royalty payments to the CFFT as described in Note 6.
9. Fair Value Measurements
The Company measures and reports certain financial instruments at fair value on a recurring basis. The Company evaluates its financial instruments subject to fair value measurements on a recurring basis to determine the appropriate level in which to classify them each reporting period. The Company determined that the warrant liability for the Series B and C Warrants, the put option on the 2014 Notes, the put option on the 2016 Notes, described further in Note 4, and the contingent consideration, described further in Note 7, were Level 3 financial instruments. The 2014 Notes and the related put option were converted into Series C Preferred Stock on December 3, 2015. The fair value of these instruments as of December 31, 2016 and December 31, 2015 was as follows (in thousands):
| Quoted Prices in Active Markets for Identical Assets (Level 1) |
Significant Other Observable Inputs (Level 2) |
Significant Unobservable Inputs (Level 3) |
||||||||||
| As of December 31, 2016: |
||||||||||||
| Put option |
$ | — | $ | — | $ | 979 | ||||||
| Warrant liability |
$ | — | $ | — | $ | 303 | ||||||
| Contingent consideration |
$ | — | $ | — | $ | 9,708 | ||||||
| As of December 31, 2015: |
||||||||||||
| Put option |
$ | — | $ | — | $ | — | ||||||
| Warrant liability |
$ | — | $ | — | $ | 274 | ||||||
122
Table of Contents
The estimated fair value of the put option on the 2014 Notes and the put option on the 2016 Notes was determined using a multi-scenario probability weighted average method analysis in which the future probability of the equity financing event was weighted for its respective probability. The Company used the following assumptions to value the put option on the 2016 Notes as of December 31, 2016. As discussed above, the 2014 Notes and the related put option were converted into Series C Preferred Stock on December 3, 2015. Therefore, there was no put option on the 2014 Notes outstanding at December 31, 2015.
| Assumption |
December 31, 2016 |
|||
| Discount rate |
0.43 | % | ||
| Probability of event |
85 | % | ||
Changes in the unobservable inputs noted above would impact the fair value of the put option and have a corresponding impact on the Company’s net loss. The probability of the automatic conversion feature was determined by management based on its consideration of the expected timeline for the next round of financing and historical experience. Increases (decreases) in discount rate would decrease (increase) the value of the put option, and an increase (decrease) in the probability of the equity financing event occurring would increase (decrease) the value of the put option.
The estimated fair value of the warrant liability (Series B and Series C warrants) was determined using a Norren Wolfson option pricing model. The assumptions used in valuing these warrants are presented in the table below.
| Assumption |
December 31, 2016 | December 31, 2015 | ||
| Expected term |
0.42 – 4.50 | 1.42 | ||
| Expected dividend yield |
— % | — % | ||
| Expected volatility |
44.65% - 60.66% | 45.33% | ||
| Risk-free interest rate |
0.58% - 1.82% | 0.84% |
Changes in the unobservable inputs noted above would impact the fair value of the liabilities and have a corresponding impact on the Company’s net loss. Increases (decreases) in the expected term and expected volatility would increase (decrease) net loss and the value of the warrant liability and an increase (decrease) in the risk-free interest rate would decrease (increase) net loss and the value of the warrant liability.
The Company did not transfer any assets measured at fair value on a recurring basis to or from Level 1 and Level 2 during the years ended December 31, 2016 and 2015.
The following table sets forth a summary of the changes in the fair value of the Company’s Level 3 financial instrument (in thousands) for the year ended December 31, 2016 and 2015:
| Warrant Liability |
Put Option on 2014 Note |
Put Option on 2016 Note |
Contingent Consideration |
|||||||||||||
| As of December 31, 2013: |
$ | 153 | $ | — | $ | — | $ | — | ||||||||
| Put option at issuance of convertible notes |
— | 2,562 | — | — | ||||||||||||
| Change in fair value |
— | 2 | — | — | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| As of December 31, 2014: |
153 | 2,564 | — | — | ||||||||||||
| Change in fair value |
121 | 144 | — | — | ||||||||||||
| Extinguishment of put option |
— | (2,708 | ) | — | — | |||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| As of December 31, 2015: |
274 | — | — | — | ||||||||||||
| Put option at issuance of 2016 Notes |
— | — | 977 | — | ||||||||||||
| Contingent consideration (see Note 7) |
— | — | — | 9,524 | ||||||||||||
| Issuance of Series C Warrants |
259 | — | — | — | ||||||||||||
| Change in fair value |
(230 | ) | 2 | 184 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Balance at December 31, 2016 |
$ | 303 | $ | — | $ | 979 | $ | 9,708 | ||||||||
|
|
|
|
|
|
|
|
|
|||||||||
123
Table of Contents
10. Redeemable Convertible Preferred Stock
The following table summarizes the Company’s redeemable convertible preferred stock as of December 31, 2016 (in thousands, except share and per share amounts).
| Redeemable Convertible Preferred Stock |
Par Value |
Authorized Shares |
Shares Issued and Outstanding |
Carrying Value |
Liquidation Value |
|||||||||||||||
| Series A |
$ | .001 | 1,799,906 | 1,799,906 | $ | 3,232 | $ | 3,254 | ||||||||||||
| Series B |
$ | .001 | 6,000,000 | 5,675,387 | $ | 17,301 | $ | 17,762 | ||||||||||||
| Series C |
$ | .001 | 8,000,000 | 4,452,582 | $ | 23,328 | $ | 23,423 | ||||||||||||
On December 3, 2015, the Company issued 1,432,482 shares of Series C Preferred Stock for net proceeds of $7,404,000, as well as an additional 2,615,308 shares of Series C Preferred Stock with a fair value of $13,758,000 related to the conversion of the 2014 Notes under the Automatic Conversion feature (See Note 4). The Company also collected an additional $1,368,000 in cash prior to December 31, 2015 for subscriptions to the Series C Preferred Stock that were issued as part of the final closing in February 2016. Management concluded that it was appropriate to present the receipt of these proceeds in equity as the investors signed individual subscription agreements and all terms and conditions of the final closing were executed and approved by the shareholders before the balance sheet date. The shares of Series C Preferred Stock related to subscriptions that were issued as part of the final closing are not presented on the December 31, 2015 balance sheet or statement of changes in redeemable preferred stock and stockholders’ deficit.
The following is a summary of the Company’s Series A redeemable convertible preferred stock (“Series A Preferred Stock”), Series B Preferred Stock, and Series C Preferred Stock at December 31, 2016 and 2015 (the “Preferred Stock”):
Voting
The holders of the Preferred Stock are entitled to vote, together with the holders of common stock, on certain matters, exclusive of certain protective provisions under the Fourth Amended and Restated Certificate of Incorporation (the “Protective Provisions”), submitted to stockholders for a vote. Each preferred stockholder is entitled to the number of votes equal to the number of shares of common stock into which each preferred share is convertible at the time of such vote.
The holders of the Preferred Stock will vote, as a single class on an as converted to common stock basis, separately from the holders of common stock, on certain Protective Provisions, including but not limited to: any liquidation event, merger, consolidation or form of reorganization; any modification of the rights and privileges of the Preferred Stock so as to adversely affect the Preferred Stock; the declaration or payment of any dividend; the redemption, repurchase or other acquisition by the Company of shares of common stock; any amendment of the Certificate of Incorporation or By-Laws of the Company; any increase in the number of authorized shares of Preferred Stock or common stock; the incurrence of certain indebtedness; and any change in the number of members of the of Board of Directors.
Dividends
The holders of Preferred Stock are entitled to receive dividends, when and if declared by the Board of Directors and out of funds legally available, at an annual rate of $0.14462 per share for the Series A Preferred Stock, $0.2504 per share for the Series B Preferred Stock, and $0.42084 per share for the Series C Preferred Stock. Dividends on the Preferred Stock are non-cumulative. As of December 31, 2016, no dividends had been declared or paid by the Company.
124
Table of Contents
Liquidation
In the event of any voluntary or involuntary liquidation, dissolution or winding up of the affairs of the Company, the holders of the Series A Preferred Stock, Series B Preferred Stock, and Series C Preferred Stock shall receive on a per share basis $1.80783, $3.12959, and $5.2605 (as adjusted), respectively, plus all declared but unpaid dividends, payable in preference and priority to any payments made to the holders of the common stock. Once paid, the remaining assets available will be distributed ratably between Preferred and common stockholders until the holders of the Series A Preferred Stock, Series B Preferred Stock, and Series C Preferred Stock receive an amount equal to two times their liquidation preference, then ratably among the common stockholders. As of December 31, 2016, the liquidation preference was $3,253,924, $17,761,634 and $23,422,808 for the Series A Preferred Stock, Series B Preferred Stock, and Series C Preferred Stock, respectively.
Conversion
Each share of Series A Preferred Stock, Series B Preferred Stock, and Series C Preferred Stock is convertible at the option of the holders at any time after the date of issuance into a number of shares of common stock as determined by dividing $1.80783, $3.12959, and $5.2605, respectively, by the conversion price in effect at the time of conversion. The conversion price of the Series A Preferred Stock, Series B Preferred Stock, and Series C Preferred Stock is $1.80783 and $3.12959, and $5.2605, respectively, and is subject to adjustment, as defined. Conversion is automatic upon the earlier of 1) the Company’s sale of common stock in a firm commitment underwritten public offering, or 2) the date specified by written consent or agreement of the holders of at least a majority of the outstanding shares of Preferred Stock.
Redemption
The Preferred Stock is redeemable at the written request from the holders of at least two-thirds of the then outstanding shares of Preferred Stock, at any time after the October 31, 2022, in three equal annual installments. The redemption price will be an amount per share equal to $1.80783, $3.12959, and $5.2605 for Series A Preferred Stock, Series B Preferred Stock, and Series C Preferred Stock, respectively, plus all declared and unpaid dividends thereon.
11. Common Stock
The Company is authorized to issue 27,000,000 shares of common stock with a par value of $0.001 per share. The following is a summary of the Company’s common stock at December 31, 2016 and 2015.
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Common stock authorized |
27,000,000 | 20,000,000 | ||||||
| Common stock outstanding |
5,396,883 | 2,041,552 | ||||||
During the years ended December 31, 2016 and, 2015, the Company issued 0 and 50,000 shares of restricted common stock, respectively. See Note 12 for disclosures related to the valuation of the Company’s common stock.
Liquidation Rights
In the event of any liquidation or dissolution of the Company, the holders of common stock are entitled to the remaining assets of the Company legally available for distribution after the payment of the full liquidation preference for all series of outstanding redeemable convertible preferred stock.
125
Table of Contents
Dividend and Voting Rights
The holders of common stock are entitled to receive dividends if and when declared by the Company, but not until all dividends on redeemable convertible preferred stock have been either (i) paid or (ii) declared and the Company has set aside funds to pay those dividends declared. Holders of common stock have the right to one vote per share.
Common Stock Reserved for Issuance
The Company’s shares of common stock reserved for issuance as of December 31, 2016 were as follows:
| 2016 | ||||
| Series A Preferred Stock |
1,799,906 | |||
| Series B Preferred Stock |
5,675,387 | |||
| Series C Preferred Stock |
4,452,582 | |||
| Series B Warrants |
289,966 | |||
| Series C Warrants |
125,885 | |||
| Stock options outstanding |
3,096,665 | |||
|
|
|
|||
| Total shares reserved |
15,440,391 | |||
|
|
|
|||
12. Stock-Based Compensation
The Company adopted the Savara Inc. Stock Option Plan (the “Plan”), pursuant to which the Company has reserved 5,300,076 shares for issuance to employees, directors, and consultants. The Plan includes 1) the option grant program providing for both incentive and non-qualified stock options, as defined by the Internal Revenue Code, and 2) the stock issuance program providing for the issuance of awards that are valued based upon common stock, including restricted stock, dividend equivalents, stock appreciation rights, phantom stock, and performance units. The Plan also allows eligible persons to purchase shares of common stock at an amount determined by the Plan Administrator. Upon a participant’s termination, the Company retains the right to repurchase unvested shares issued in conjunction with the stock issuance program at the fair market value per share as of the date of termination.
To date the Company has issued incentive and non-qualified options and restricted stock to employees and non-employees under the Plan. The terms of the stock options, including the exercise price per share and vesting provisions, are determined by the Board of Directors. Stock options are granted at exercise prices not less than the estimated fair market value of the Company’s common stock at the date of grant based upon numerous objective and subjective factors including: third-party valuations, preferred stock transactions with third parties, current operating and financial performance, management estimates and future expectations. Stock option grants typically vest quarterly over three to four years and expire ten years from the grant date, and restricted stock grants vest on a quarterly basis over four years and expire ten years from the grant date. Inception to date, the Company has issued 992,563 shares of restricted stock.
Stock-based compensation expense is included in the following line items in the accompanying statements of operations and comprehensive loss for the years ended December 31 (in thousands):
| 2016 | 2015 | 2014 | ||||||||||
| Research and development |
$ | 93 | $ | 69 | $ | 42 | ||||||
| Selling, general and administrative |
116 | 84 | 99 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total stock-based compensation |
$ | 209 | $ | 153 | $ | 141 | ||||||
|
|
|
|
|
|
|
|||||||
126
Table of Contents
Stock Options
The Company values stock options using the Black-Scholes option-pricing model, which requires the input of subjective assumptions, including the risk-free interest rate, expected life, expected stock price volatility and dividend yield. The risk-free interest rate assumption is based upon observed interest rates for constant maturity U.S. Treasury securities consistent with the expected term of the Company’s employee stock options. The expected life represents the period of time the stock options are expected to be outstanding and is based on the simplified method. The Company uses the simplified method due to the lack of sufficient historical exercise data to provide a reasonable basis upon which to otherwise estimate the expected life of the stock options. Expected volatility is based on historical volatilities for publicly traded stock of comparable companies over the estimated expected life of the stock options. The Company assumes no dividend yield because dividends are not expected to be paid in the near future, which is consistent with the Company’s history of not paying dividends.
The following table summarizes the assumptions used for estimating the fair value of stock options granted to employees for the years ended December 31:
| 2016 | 2015 | 2014 | ||||
| Risk-free interest rate |
1.49% - 2.31% | 1.83% - 1.91% | 1.70% - 1.91% | |||
| Expected term (years) |
6.00 - 6.25 | 5.96 - 6.08 | 5.81 - 6.06 | |||
| Expected volatility |
50%-63% | 45%-50% | 78%-81% | |||
| Dividend yield |
— % | — % | — % |
The following table summarizes the assumptions used for estimating the fair value of stock options granted to non-employees for the year ended December 31:
| 2016 | 2015 | 2014 | ||||
| Risk-free interest rate |
2.31% - 2.38% | 1.90% - 2.27% | 2.14% - 2.26% | |||
| Expected term (years) |
9.57 - 9.76 | 9.57 - 9.76 | 9.57 - 9.76 | |||
| Expected volatility |
63% | 50% | 77% | |||
| Dividend yield |
— % | — % | — % |
The following table summarizes the stock option activity for employees and non-employees for the year ended December 31, 2016:
| Number of Options |
Weighted Average Exercise Price |
Weighted Average Remaining Contractual Term |
Aggregate Intrinsic Value |
|||||||||||||
| (in years) | ||||||||||||||||
| Outstanding balance at December 31, 2015 |
1,842,455 | $ | 0.60 | 8.45 | $ | — | ||||||||||
| Granted |
1,576,929 | 0.98 | 6.06 | — | ||||||||||||
| Exercised |
(1,406 | ) | 0.48 | — | — | |||||||||||
| Cancelled |
(321,313 | ) | 0.85 | — | — | |||||||||||
|
|
|
|||||||||||||||
| Outstanding balance at December 31, 2016 |
3.096,665 | $ | 0.78 | 8.54 | $ | 746,647 | ||||||||||
|
|
|
|||||||||||||||
| Exercisable at December 31, 2016 |
1,013,650 | $ | 0.47 | 6.49 | $ | 564,326 | ||||||||||
|
|
|
|||||||||||||||
| Vested and expected to vest at December 31, 2016 |
3,096,665 | $ | 0.78 | 8.54 | $ | 774,647 | ||||||||||
|
|
|
|||||||||||||||
127
Table of Contents
The following table provides a summary of options issued to employees and non-employees that are outstanding and vested as of December 31, 2016:
| Exercise Prices | Number Outstanding |
Weighted- Average Life (in Years) |
Number Exercisable |
Weighted- Average Life (in Years) |
||||||||||||||
| $ | 0.11 | 47,230 | 2.08 | 47,230 | 2.08 | |||||||||||||
| $ | 0.18 | 12 | 2.95 | 12 | 2.95 | |||||||||||||
| $ | 0.30 | 86,437 | 4.09 | 86,437 | 4.09 | |||||||||||||
| $ | 0.32 | 60,000 | 4.95 | 60,000 | 4.95 | |||||||||||||
| $ | 0.38 | 523,276 | 6.51 | 440,389 | 6.24 | |||||||||||||
| $ | 0.48 | 178,000 | 7.00 | 163,750 | 6.99 | |||||||||||||
| $ | 0.85 | 860,000 | 8.97 | 215,832 | 8.97 | |||||||||||||
| $ | 0.88 | 217,710 | 9.82 | — | — | |||||||||||||
| $ | 1.03 | 1,124,000 | 9.96 | — | — | |||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||
| 3,096,665 | 8.54 | 1,013,650 | 6.49 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||
The weighted average grant date fair values for the Company’s stock options granted during the years ended December 31, 2016, 2015 and 2014 were $0.98 per share, $0.85 per share and $0.27, respectively. The total compensation cost related to non-vested stock options not yet recognized as of December 31, 2016 was $1,087,274, which will be recognized over a weighted average period of approximately 8.54 years. 1,406, 0 and 0 stock options were exercised during the years ended December 31, 2016, 2015 and 2014, respectively.
During the year ended December 31, 2016, the Company granted options to purchase a total of 10,000 shares of common stock to non-employees under the Plan.
The Company recorded stock-based compensation expense for options issued to non-employees of $3,400, $4,000 and $10,000 for the years ended December 31, 2016, 2015 and 2014, respectively. As of December 31, 2016, 104,159 non-employee options were vested and outstanding.
Restricted Stock
The Company values stock-based compensation related to grants of its restricted stock based on the fair value of the Company’s common stock as of the grant date and recognizes the expense over the requisite service period, usually four years, adjusted for estimated forfeitures. To determine the value of its common stock, the Company utilized the Option Pricing Method. The valuation methodology includes estimates and assumptions that require the Company’s judgment. Inputs used to determine the estimated fair value of the Company’s common stock include the equity value of the Company, expected timing to a liquidity event of 2.0-2.5 years, a risk-free interest rate of 0.58%-0.59% and the expected volatility of 45%-66%. Generally, increases or decreases in these unobservable inputs would result in a directionally similar impact to the fair value measurement of the Company’s common stock. During the year ended December 31, 2016, the Company did not issue any shares of restricted stock to employees for compensation. The Company recorded stock-based compensation expense related to the restricted stock of $53,000, $60,000 and $57,000 for the years ended December 31, 2016, 2015 and 2014, respectively.
128
Table of Contents
The following table summarizes the restricted share activity for the years ended December 31, 2016 and 2015:
| Restricted Shares |
Weighted- Average Grant Date Fair Value |
|||||||
| Nonvested at December 31, 2013 |
398,063 | $ | 0.41 | |||||
| Granted |
165,776 | 0.38 | ||||||
| Vested |
(112,711 | ) | 0.42 | |||||
|
|
|
|
|
|||||
| Nonvested at December 31, 2014 |
451,128 | $ | 0.41 | |||||
| Granted |
50,000 | 0.47 | ||||||
| Vested |
(192,572 | ) | 0.41 | |||||
|
|
|
|
|
|||||
| Nonvested at December 31, 2015 |
308,556 | 0.44 | ||||||
| Granted |
— | — | ||||||
| Vested |
(123,556 | ) | 0.43 | |||||
|
|
|
|
|
|||||
| Nonvested at December 31, 2016 |
185,000 | $ | 0.40 | |||||
|
|
|
|
|
|||||
The total fair value of restricted stock that vested during the year ended December 31, 2016 was $27,000. The total compensation cost related to non-vested restricted stock not yet recognized as of December 31, 2016 was $66,849, which will be recognized over a weighted average period of approximately 1.55 years.
13. Income Taxes
The components of loss before income taxes for the years ended December 31, 2016 and 2015 are as follows (in thousands):
| 2016 | 2015 | 2014 | ||||||||||
| Domestic |
$ | (8,502 | ) | $ | (8,999 | ) | $ | (6,282 | ) | |||
| Foreign |
(2,778 | ) | — | — | ||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | (11,280 | ) | $ | (8,999 | ) | $ | (6,282 | ) | |||
|
|
|
|
|
|
|
|||||||
The Company recorded no federal provision for income taxes for the years ended December 31, 2016, 2015 and 2014 due to reported net losses in each year. The Company recorded no state provision for income taxes for the years ended December 31, 2016, 2015 and 2014, due to revenues below the minimum tax threshold. The Company recorded a foreign current income tax benefit of approximately $357,000 related to the refundable research credit.
A reconciliation of the expected income tax expense (benefit) computed using the federal statutory income tax rate to the Company’s effective income tax rate is as follows for the years ended December 31, 2016 and 2015 (in thousands):
| 2016 | 2015 | 2014 | ||||||||||
| Income Tax Expense (Benefit) Computed at Federal Statutory Tax Rate |
$ | (3,835 | ) | $ | (3,059 | ) | $ | (2,136 | ) | |||
| Change in Valuation Allowance |
4,009 | 3,488 | 3,421 | |||||||||
| Orphan Drug & Research Credits Generated |
(2,435 | ) | (1,983 | ) | (2,285 | ) | ||||||
| Orphan Drug & Research Credit Expense Disallowance |
1,126 | 674 | 777 | |||||||||
| State Research Credits Generated, Net of Federal Benefit |
— | (175 | ) | (95 | ) | |||||||
| Impact of Foreign Operations |
333 | — | — | |||||||||
| Interest on Convertible Debt |
— | 867 | 284 | |||||||||
| Other Permanent Differences |
445 | 188 | 34 | |||||||||
|
|
|
|
|
|
|
|||||||
| Total |
$ | (357 | ) | $ | — | $ | — | |||||
|
|
|
|
|
|
|
|||||||
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company has established a valuation allowance due to uncertainties regarding the realization of deferred tax assets based upon
129
Table of Contents
the Company’s lack of earnings history. During the years ended December 31, 2016 and December 31, 2015, the valuation allowance increased by $4.0 million and $3.5 million, respectively. Significant components of the Company’s deferred tax assets and liabilities are as follows (in thousands):
| December 31, | ||||||||
| 2016 | 2015 | |||||||
| Deferred Tax Liabilities: |
||||||||
| Stock-based Compensation |
$ | 2 | $ | 27 | ||||
| Depreciation |
257 | — | ||||||
| Intangible Assets |
2,305 | — | ||||||
|
|
|
|
|
|||||
| Total Deferred Tax Liabilities |
2,564 | 27 | ||||||
| Deferred Tax Assets: |
||||||||
| Net Operating Loss Carryforwards |
7,483 | 5,672 | ||||||
| Amortization |
11 | 6 | ||||||
| Credit Carryforwards |
7,241 | 5,190 | ||||||
| Charitable Contributions |
— | 81 | ||||||
| Accrued Liabilities & Other |
455 | — | ||||||
|
|
|
|
|
|||||
| Total Deferred Tax Assets |
15,190 | 10,949 | ||||||
|
|
|
|
|
|||||
| Subtotal |
12,626 | 10,922 | ||||||
| Valuation Allowance |
(14,931 | ) | (10,922 | ) | ||||
|
|
|
|
|
|||||
| Net Deferred Taxes |
$ | (2,305 | ) | $ | — | |||
|
|
|
|
|
|||||
Given the significant risk associated with the completion and commercialization of the Company’s products that will be derived from the indefinite lived in-process research and development asset, management is not considering the corresponding deferred tax liability as a source of income for purposes of its valuation allowance due to the uncertainty of if and when this temporary difference would ever reverse. As of December 31, 2016, 2015 and 2014, the Company had net operating loss (“NOL”) carryforwards for federal income tax purposes of approximately $21.7 million, $16.7 million and $12.8 million, respectively. The Company also had available research and orphan drug tax credit carryforwards for federal income tax purposes of approximately $7.2 million, $5.0 million and $3.0 million, respectively. If not utilized, these carryforwards expire at various dates beginning in 2028. As of December 31, 2016, 2015 and 2014, the Company had state research and development tax credit carryforwards of approximately $69,000, $300,000 and $100,000, respectively, which will begin to expire in 2034 if not utilized. As of December 31, 2016, 2015 and 2014, the Company had foreign NOL carryforwards of approximately $0.4 million, $0 and $0, respectively, which have an indefinite carryforward period.
Utilization of the NOL and tax credit carryforwards may be subject to a substantial annual limitation due to ownership change limitations that have occurred previously or that could occur in the future, as provided by Section 382 of the Internal Revenue Code of 1986 (“Section 382”), as well as similar state provisions. Ownership changes may limit the amount of NOL carryforwards and tax credit carryforwards that can be utilized annually to offset future taxable income and tax, respectively. In general, an ownership change, as defined by Section 382, results from transactions that increase the ownership of 5% shareholders in the stock of a corporation by more than 50 percentage points in the aggregate over a three-year period. The Company has not performed a study to determine whether any ownership change has occurred since the Company’s formation through December 31, 2016. However, the Company believes that it has experienced at least one ownership change in the past and that it may experience additional ownership changes as a result of subsequent shifts in its stock ownership. Should there be an ownership change that has occurred or will occur, the Company’s ability to utilize existing carryforwards could be substantially restricted and may result in the expiration of such carryforwards prior to utilization.
The Company applies the accounting guidance in ASC 740 related to accounting for uncertainty in income taxes. The Company’s reserves related to taxes are based on a determination of whether, and how much of, a tax benefit taken by the Company in its tax filings or positions is more likely than not to be realized following resolution of any potential contingencies present related to the tax benefit. As of December 31, 2016, 2015 and 2014, the Company had no unrecognized tax benefits. During the years ended December 31, 2016, 2015 and 2014, the Company had no interest and penalties related to income taxes.
130
Table of Contents
The Company files income tax returns in the U.S. federal, Texas, and foreign jurisdictions. As of December 31, 2016, the statute of limitations for assessment by the Internal Revenue Service (“IRS”) is open for the 2013 and subsequent tax years, although carryforward attributes that were generated for tax years prior to then may still be adjusted upon examination by the IRS if they either have been, or will be, used in a future period. The 2012 and subsequent tax years remain open and subject to examination by the State of Texas. The 2016 tax years remain open and subject to examination by the foreign taxing authorities. There are currently no federal, state, or foreign income tax audits in progress.
14. Net Loss per Share
Basic net loss per share is computed by dividing the net loss by the weighted-average number of common shares outstanding. Diluted net loss per share is computed similarly to basic net loss per share except that the denominator is increased to include the number of additional common shares that would have been outstanding if the potential common shares had been issued and if the additional common shares were dilutive. Diluted net loss per share is the same as basic net loss per common share, since the effects of potentially dilutive securities are antidilutive.
As of December 31, 2016, and 2015, potentially dilutive securities include:
| December 31, 2016 | December 31, 2015 | |||||||
| Awards under equity incentive plan |
3,096,665 | 1,842,455 | ||||||
| Unvested restricted shares |
185,000 | 308,556 | ||||||
| Series A Contingent Redeemable Preferred Stock |
1,799,906 | 1,799,906 | ||||||
| Series B Contingent Redeemable Preferred Stock |
5,675,387 | 5,675,387 | ||||||
| Series C Contingent Redeemable Preferred Stock |
4,452,582 | 4,038,790 | ||||||
| 2016 Series C Convertible Note |
869,409 | — | ||||||
| Warrants to purchase Series B Contingent Redeemable Preferred Stock |
289,966 | 289,966 | ||||||
| Warrants to purchase Series C Contingent Redeemable Preferred Stock |
125,885 | — | ||||||
|
|
|
|
|
|||||
| Total |
16,494,800 | 13,955,060 | ||||||
|
|
|
|
|
|||||
The following table reconciles basic earnings per share of common stock to diluted earnings per share of common stock for the years ended December 31, 2016, 2015, and 2014.
| December 31, 2016 | December 31, 2015 | December 31, 2014 | ||||||||||
| Net loss |
$ | (10,923 | ) | $ | (8,999 | ) | $ | (6,282 | ) | |||
| Accretion of redeemable convertible preferred stock |
(94 | ) | (183 | ) | (121 | ) | ||||||
|
|
|
|
|
|
|
|||||||
| Net loss attributable to common stockholders |
(11,017 | ) | (9,182 | ) | (6,403 | ) | ||||||
| Undistributed earnings and loss available to common stockholders |
(11,017 | ) | (9,182 | ) | (6,403 | ) | ||||||
| Weighted average common shares outstanding, basic and diluted |
3,348,647 | 1,653,259 | 1,503,058 | |||||||||
| Basic and diluted EPS |
$ | (3.29 | ) | $ | (5.55 | ) | $ | (4.26 | ) | |||
|
|
|
|
|
|
|
|||||||
15. Subsequent Events
The Company has evaluated subsequent events through March 10, 2017, the date the financial statements were available to be issued.
Merger Agreement with Mast Therapeutics
On January 6, 2017, Savara entered into an Agreement and Plan of Merger and Reorganization, or the Merger Agreement, with Mast Therapeutics, Inc., or Mast, a publicly traded company on the NYSE MKT (MSTX), pursuant to which, among other things, subject to the satisfaction or waiver of the conditions set forth in the Merger Agreement, a wholly owned subsidiary of Mast will merge with and into Savara, with Savara becoming a wholly-owned subsidiary of Mast and the surviving corporation of the Merger. Mast is a biopharmaceutical company focused on developing clinical-stage therapies for serious or life-threatening diseases. At the closing of
131
Table of Contents
the Merger, each outstanding share of Savara’s common stock will be converted into the right to receive shares of common stock (to be determined at a later date) of Mast as well as the payment of cash in lieu of fractional shares. Immediately following the effective time of the Merger, Mast equity holders are expected to own approximately 23% of the combined company, with Savara’s preexisting equity holders expected to own approximately 77%. At the time the financial statements were available to be issued, the valuation of the Company’s common stock as well as the initial accounting for the business combination was incomplete. As a result, additional disclosures related to the Merger with Mast are unavailable.
Modification of the 2016 Notes
The 2016 Notes and the Warrants were amended to include a conversion clause in the case of a reverse merger with Mast, further discussed above. The amendment provides the warrant holder the right to voluntarily exercise the Warrants; however, the 2016 Notes are automatically converted in the case of a reverse merger with Mast. Notes that were issued on or prior to August 15, 2016 were assigned a conversion price of $4.22 and notes that were issued after August 15, 2016, were assigned a conversion price of 80% of the amount equal to the average trading price of Mast’s common stock for the twenty day period ending two days prior to the closing of the acquisition of Mast by the Company, as adjusted by the exchange ratio described in the Merger Agreement.
Events Subsequent to the Original Issuance of Financial Statements (Unaudited)
On April 27, 2017, the merger with Mast was consummated. In connection with the reissuance of the financial statements, the Company has evaluated subsequent events through April 27, 2017, the date the financial statements were available to be reissued.
On April 27, 2017, immediately prior to the effective time of the merger, Mast amended and restated its certificate of incorporation to effect a reverse stock split and change its name to “Savara Inc.” The amendment and restatement of Mast’s certificate of incorporation was approved by Mast’s stockholders at a special meeting of stockholders on April 27, 2017.
In connection with the merger with Mast, the Company became a wholly-owned subsidiary of Mast and changed its name to Aravas Inc.
132
Table of Contents
To the Board of Directors of Savara Inc.
We have audited the accompanying consolidated financial statements of Serendex Pharmaceuticals A/S and subsidiaries, which comprise the consolidated balance sheets as of December 31, 2015 and 2014, and the related consolidated income statement, statement of comprehensive income, cash flows and changes in equity for the years then ended, and the related notes to the financial statements.
Management’s responsibility for the financial statements
Management is responsible for the preparation and fair presentation of these consolidated financial statements in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board; this includes the design, implementation, and maintenance of internal control relevant to the preparation and fair presentation of consolidated financial statements that are free from material misstatement, whether due to fraud or error.
Auditor’s responsibility
Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We conducted our audits in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free from material misstatement.
An audit involves performing procedures to obtain audit evidence about the amounts and disclosures in the consolidated financial statements. The procedures selected depend on the auditor’s judgment, including the assessment of the risks of material misstatement of the consolidated financial statements, whether due to fraud or error. In making those risk assessments, the auditor considers internal control relevant to the entity’s preparation and fair presentation of the consolidated financial statements in order to design audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the entity’s internal control. Accordingly, we express no such opinion. An audit also includes evaluating the appropriateness of accounting policies used and the reasonableness of significant accounting estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements.
We believe that the audit evidence we have obtained is sufficient and appropriate to provide a basis for our audit opinion.
Opinion
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Serendex Pharmaceutical A/S and subsidiaries as of December 31, 2015 and 2014, and the results of their operations and their cash flows for the years then ended in accordance International Financial Reporting Standards as issued by the International Accounting Standards Board.
/s/ Grant Thornton
GRANT THORNTON
Statsautoriseret Revisionspartnerselskab
Copenhagen, Denmark
February 7, 2017
133
Table of Contents
for the period 1 January — 31 December
| DKK thousand |
Notes | 2015 | 2014 | |||||||||
| Net revenue |
3 | 153 | 786 | |||||||||
| Cost of goods sold |
(125 | ) | (326 | ) | ||||||||
|
|
|
|
|
|||||||||
| Gross profit |
27 | 461 | ||||||||||
|
|
|
|
|
|||||||||
| Staff expenses |
4, 5 | (18,233 | ) | (13,590 | ) | |||||||
| External expenses |
5 | (8,542 | ) | (11,537 | ) | |||||||
| Other expenses |
5 | (7,282 | ) | (900 | ) | |||||||
|
|
|
|
|
|||||||||
| Operating profit/loss (-) |
(34,030 | ) | (25,566 | ) | ||||||||
|
|
|
|
|
|||||||||
| Net financials |
7 | (3,182 | ) | (3,787 | ) | |||||||
|
|
|
|
|
|||||||||
| Profit/loss (-) before tax |
(37,212 | ) | (29,353 | ) | ||||||||
|
|
|
|
|
|||||||||
| Tax expenses |
8 | 3,359 | 7,202 | |||||||||
|
|
|
|
|
|||||||||
| Net profit/loss (-) |
(33,853 | ) | (22,151 | ) | ||||||||
|
|
|
|
|
|||||||||
STATEMENT OF COMPREHENSIVE INCOME
| DKK thousand |
Notes | 2015 | 2014 | |||||||||
| Net profit/loss (-) |
(33,853 | ) | (22,151 | ) | ||||||||
| Other comprehensive income |
0 | 0 | ||||||||||
|
|
|
|
|
|||||||||
| Total comprehensive income |
(33,853 | ) | (22,151 | ) | ||||||||
|
|
|
|
|
|||||||||
| Total earnings per share (DKK) |
9 | (2.25 | ) | (1.47 | ) | |||||||
| Diluted total earnings per share (DKK) |
9 | (2.21 | ) | (1.47 | ) | |||||||
134
Table of Contents
AT 31 DECEMBER
ASSETS
| DKK thousand |
Notes | 2015 | 2014 | |||||||||
| NON-CURRENT ASSETS |
||||||||||||
| Intangible assets |
||||||||||||
| Development projects |
11 | 58,763 | 29,417 | |||||||||
| Tangible assets |
||||||||||||
| Plant and equipment |
12 | 219 | 278 | |||||||||
| Financial assets |
||||||||||||
| Long-term deferred tax |
14 | 0 | 2,516 | |||||||||
| Non-current receivables |
||||||||||||
| Deposits |
13 | 199 | 194 | |||||||||
|
|
|
|
|
|||||||||
| Total non-current assets |
59,182 | 32,406 | ||||||||||
|
|
|
|
|
|||||||||
| CURRENT ASSETS |
||||||||||||
| Inventories |
15 | 1,244 | 1,249 | |||||||||
| Receivables |
||||||||||||
| Trade receivables |
0 | 619 | ||||||||||
| Tax receivables |
14 | 5,875 | 6,250 | |||||||||
| Prepayments |
0 | 750 | ||||||||||
| Other receivables |
1,117 | 566 | ||||||||||
|
|
|
|
|
|||||||||
| Total receivables |
6,992 | 8,185 | ||||||||||
|
|
|
|
|
|||||||||
| Cash and cash equivalents |
5,974 | 20,460 | ||||||||||
| Total current assets |
14,210 | 29,893 | ||||||||||
|
|
|
|
|
|||||||||
| Total assets |
73,391 | 62,299 | ||||||||||
|
|
|
|
|
|||||||||
| EQUITY AND LIABILITIES |
||||||||||||
| EQUITY |
||||||||||||
| Share capital |
12,134 | 1,506 | ||||||||||
| Retained earnings |
49,059 | 32,179 | ||||||||||
|
|
|
|
|
|||||||||
| Total equity |
61,193 | 33,685 | ||||||||||
|
|
|
|
|
|||||||||
| LIABILITIES |
||||||||||||
| Non-current liabilities |
||||||||||||
| Long-term loans from shareholders and management |
16 | 1,168 | 23,464 | |||||||||
|
|
|
|
|
|||||||||
| Total non-current liabilities |
1,168 | 23,464 | ||||||||||
|
|
|
|
|
|||||||||
| Current liabilities |
||||||||||||
| Trade payables |
5,921 | 386 | ||||||||||
| Other current liabilities |
5,108 | 4,763 | ||||||||||
|
|
|
|
|
|||||||||
| Total current liabilities |
11,029 | 5,149 | ||||||||||
|
|
|
|
|
|||||||||
| Total liabilities |
12,197 | 28,614 | ||||||||||
|
|
|
|
|
|||||||||
| Total equity and liabilities |
73,391 | 62,299 | ||||||||||
|
|
|
|
|
|||||||||
135
Table of Contents
Consolidated Changes in equity
| DKK thousand |
Notes | Share capital | Retained earnings |
Total | ||||||||||||
| Equity at 1 January 2014 |
1,144 | 4,974 | 6,118 | |||||||||||||
| Profit/loss (-) |
0 | (22,151 | ) | (22,151 | ) | |||||||||||
| Other comprehensive income |
0 | 0 | 0 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total comprehensive income |
1,144 | (17,177 | ) | (16,033 | ) | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Share capital increase |
362 | 0 | 362 | |||||||||||||
| Share premium by IPO in 2014 |
0 | 56,647 | 56,647 | |||||||||||||
| Capital transactions costs |
0 | (7,291 | ) | (7,291 | ) | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Equity at 31 December 2014 |
1,506 | 32,179 | 33,685 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Equity at 1 January 2015 |
1,506 | 32,179 | 33,685 | |||||||||||||
| Profit/loss (-) |
0 | (33,853 | ) | (33,853 | ) | |||||||||||
| Other comprehensive income |
0 | 0 | 0 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total comprehensive income |
0 | (33,853 | ) | (33,853 | ) | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Share-based incentive |
4 | 0 | 651 | 651 | ||||||||||||
| Share capital increase |
10,628 | 0 | 10,628 | |||||||||||||
| Share premium by converted shareholder loan |
0 | 51,394 | 51,394 | |||||||||||||
| Capital transactions costs |
0 | (1,311 | ) | (1,311 | ) | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Equity at 31 December 2015 |
12,134 | 49,059 | 61,193 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
The share capital comprises of 15,055,150 shares (2014: 15,055,150 shares), each with a nominal value of DKK 0.10. shareholder loan conversion recognized end of 2015 comprises of 106,279,592 shares, also each with a nominal value of DKK 0.10. No shares hold particular rights. Conversion registered in “Erhvervsstyrelsen” on the 13.01.2016.
136
Table of Contents
Consolidated Cash Flow Statement
for the period 1 January 2015 — 31 December
| DKK thousand |
Notes | 2015 | 2014 | |||||||||
| Profit/loss (-)before tax |
(37,212 | ) | (29,353 | ) | ||||||||
| Adjustments |
21 | 9,322 | 4,138 | |||||||||
| Change in working capital |
22 | 6,703 | 450 | |||||||||
| Cash flow from operating activities before net financials |
(21,187 | ) | (24,765 | ) | ||||||||
| Currency gain/loss |
7 | 796 | (1,315 | ) | ||||||||
| Net interest costs |
7 | (3,978 | ) | (2,472 | ) | |||||||
| Interest accrued on shareholder loan |
3,725 | 0 | ||||||||||
| Cash flow from ordinary activities |
(20,644 | ) | (28,552 | ) | ||||||||
| Tax reimbursement |
14 | 6,250 | 1,250 | |||||||||
|
|
|
|
|
|||||||||
| Cash flow from operating activities |
(14,394 | ) | (27,302 | ) | ||||||||
|
|
|
|
|
|||||||||
| Addition of intangible assets |
11 | (34,776 | ) | (16,916 | ) | |||||||
| Addition of tangible assets |
12 | 0 | (313 | ) | ||||||||
| Paid deposits |
13 | (5 | ) | (194 | ) | |||||||
|
|
|
|
|
|||||||||
| Cash flow from investment activities |
(34,781 | ) | (17,422 | ) | ||||||||
|
|
|
|
|
|||||||||
| Loans received from shareholders |
36,000 | 13,439 | ||||||||||
| Share capital increase |
0 | 362 | ||||||||||
| Share premium by IPO |
0 | 56,647 | ||||||||||
| Capital transaction costs |
(1,311 | ) | (7,517 | ) | ||||||||
|
|
|
|
|
|||||||||
| Cash flow from financial activities |
34,689 | 62,931 | ||||||||||
|
|
|
|
|
|||||||||
| Cash flow in total |
(14,487 | ) | 18,206 | |||||||||
| Cash and cash equivalents at the beginning of the year |
20,460 | 2,253 | ||||||||||
|
|
|
|
|
|||||||||
| Cash and cash equivalents end of period |
5,974 | 20,460 | ||||||||||
|
|
|
|
|
|||||||||
137
Table of Contents
DKK thousand
1. CAPITAL RESOURCES
Serendex intends to license its products to pharmaceutical companies and thereby derive income from a combination of fixed payments and ongoing royalty income. Until Serendex has established such a license agreement, Serendex will be a capital-consuming company due to investments in drug development and in further strengthening of the pipeline. Therefore, it is vital that the company always has sufficient financial resources.
Serendex has a satisfactory cash situation for 2016 to continue the phase IIb clinical trial of GM-CSF for ARDS and the pivotal phase II/III clinical trial of Molgradex® for PAP according to plans. Hence, the annual report for 2015 has been prepared for on-going business.
In order to pursue the development strategy as outlaid in the Management Report, Serendex is dependent on acquiring additional capital during 2016 to continue operations in 2017 and onwards. Serendex will therefore continue to investigate opportunities and terms for entering into strategic partnerships or mergers and/or licensing agreements that will strengthen Serendex’s financial position. In addition, the company will investigate opportunities for receiving additional loans or equity financing.
2. ESTIMATES AND JUDGEMENTS
The preparation of the consolidated financial statements requires the making of estimates and judgments that affect the reporting of assets, liabilities and expenses. The estimates and judgments are reviewed on an ongoing basis. Estimates and judgments are based on historical results and on various other assumptions, which Serendex believes to be reasonable under the circumstances. However, the actual results may differ significantly from the estimates. We believe that the accounting policies relating to development costs and deferred tax involve estimates or judgments by management that could materially affect the reported financial position and results.
Development costs
Serendex is confident it will obtain approval of its pipeline products, as the products are based on an existing approved drug, and hold the evidence to support this. Further, phase IIb clinical trial of Molgradex® for ARDS and pivotal phase II/III clinical trial Molgradex® for PAP is initiated. Additionally, Serendex is confident, that it will acquire the necessary resources to either sell or complete the development. Thus, management judge that the technical feasibility criterion in IAS 38.57 is met.
The carrying amount of capitalised development costs is DKK 58.8 million for the group (2014: DKK 29.4 million).
Deferred tax
Due to tax credit reimbursement instalment by the Danish government, Serendex Group expects to be reimbursed DKK 5.9 million of the tax asset in Q4 2016 — hence the tax receivable has been recognized in the balance sheet.
The long-term deferred tax asset has been evaluated against the future income within the next three fiscal years and will not be recognised in the balance sheet, as they are not realizable.
The long-term deferred tax asset amount is DKK 5.6 million as of 31 December 2015 (2014: DKK 8.7 million).
138
Table of Contents
3. SEGMENT DATA
As of 31 December 2015, Serendex has only one segment according to IFRS. The goods sold can be categorized as follows:
| 2015 | 2014 | |||||||
| Revenue from sales of active pharmaceutical ingredient (API) |
147 | 377 | ||||||
| Revenue from named patient sales (NPS) |
0 | 410 | ||||||
| Other Sales |
6 | 0 | ||||||
|
|
|
|
|
|||||
| Total segmented revenue |
153 | 787 | ||||||
|
|
|
|
|
|||||
The entire revenue is based on major customers (>10% of the total revenue). The revenue is globally allocated as 100% in Denmark. All tangible and intangible assets are located in Denmark.
4. STAFF EXPENSES
| 2015 | 2014 | |||||||
| Staff |
||||||||
| Salaries, cash bonus, etc. |
8,627 | 6,123 | ||||||
| Pension costs or other social security costs |
1,826 | 834 | ||||||
| Share based incentive |
130 | 0 | ||||||
| Other staff costs |
398 | 79 | ||||||
|
|
|
|
|
|||||
| 10,981 | 7,036 | |||||||
|
|
|
|
|
|||||
| Management |
||||||||
| Fees to Board of Directors |
1,400 | 1,050 | ||||||
| Salary, cash bonus, etc. to Executive Management |
4,905 | 5,245 | ||||||
| Pension contributions to Executive Management |
426 | 259 | ||||||
| Share-based incentive |
521 | 0 | ||||||
|
|
|
|
|
|||||
| 7,252 | 6,554 | |||||||
|
|
|
|
|
|||||
| Total staff expenses |
18,233 | 13,590 | ||||||
|
|
|
|
|
|||||
| Full year average number of full time employees (FTE) |
12.3 | 7.5 | ||||||
| FTEs as of end of period |
12.3 | 10.8 | ||||||
Remuneration to Executive Management is based on a fixed salary and pension as well as a potential cash bonus and share-based incentive. If the majority of votes in Serendex changes hands or is transferred by agreement, or Serendex is dissolved by merger or demerger, or active transition or contractual relationship actually involves the same change in ownership or control conditions, the Board shall inform the Executive Management within 14 days after the Board becomes aware of this. Serendex termination notice to the Executive Management is then extended by nine months so that the Executive Management will be entitled to a notice period of a total of 18 months.
The increase in total staff expenses is primarily due to the increased number of employees, based on the increased activity level.
Share-based incentive
In order to encourage common and sustainable long term goals for the participants and Serendex’s shareholders in line with the company’s strategy, Serendex Pharmaceuticals A/S has established a share-based incentive plan. Thus, the Board of directors has granted warrants to the company’s management and selected employees.
139
Table of Contents
The warrants are granted in accordance with the authorisation given to the Board of Directors by the shareholders. Grant takes place at the date of establishment of the programme, allocation is subject to meeting certain milestones and exercise is by default subject to continuing employment with the company.
The terms of the share-based incentive plan are published together with the notice of the company’s General Meeting.
Assumptions for warrants granted in 2015
The fair value of the share options is calculated using Black-Scholes option pricing model. The share options were granted in April 2015 and the assumptions are shown below.
| 2015 | 2014 | |||||||
| Expected life of the warrant in years (average) |
5.0 | n.a. | ||||||
| Expected volatility (historical volatility — 12 months) |
76 | % | n.a. | |||||
| Expected dividend per share (in DKK) |
0 | n.a. | ||||||
| Risk-free interest rate (based on Danish government bonds) |
4.0 | % | n.a. | |||||
| Share price at end of period (DKK) |
0.66 | n.a. | ||||||
Outstanding warrant plans
The price and exercise period for the grant are stated in the table below.
| Outstanding warrants |
Executive Management No. of warrants |
Key employees No. of warrants |
Average exercise price DKK |
Fair value DKK ‘000 |
||||||||||||
| Outstanding at the beginning of the year |
0 | 0 | n.a | n.a | ||||||||||||
| Grant in period |
200,000 | 50,000 | 0.10 | 651 | ||||||||||||
| Exercise in period |
0 | 0 | n.a | n.a | ||||||||||||
| Fair value adjustment |
(505 | ) | ||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
| Outstanding warrants end of period |
200,000 | 50,000 | 0.10 | 146 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||
Board of directors are not eligible to participate in Serendex’s incentive programme
| Exercisable and outstanding warrants |
Issued warrants |
Exercised/ reversed |
Outstanding/ not exercised |
Exercise period |
||||||||||||
| Warrant scheme for 2015 |
250,000 | 0 | 250,000 | |
04.03.2016 (24.03.2020) |
| ||||||||||
|
|
|
|
|
|
|
|||||||||||
| Warrant scheme exercisable end of period |
250,000 | 0 | 250,000 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
5. OTHER EXPENSES AND COSTS BY FUNCTION
| 2015 | 2014 | |||||||
| Other expenses |
||||||||
| Write-down of intangible assets |
5,430 | 0 | ||||||
| Capital cost (funding) |
1,852 | 900 | ||||||
|
|
|
|
|
|||||
| 7,282 | 900 | |||||||
|
|
|
|
|
|||||
| Costs by function |
||||||||
| Sales and distribution costs |
722 | 512 | ||||||
| Expensed development costs |
6,940 | 4,200 | ||||||
| Administrative expenses |
26,395 | 21,315 | ||||||
|
|
|
|
|
|||||
| 34,057 | 26,027 | |||||||
|
|
|
|
|
|||||
140
Table of Contents
6. FEE TO STATUTORY AUDITORS
| 2015 | 2014 | |||||||
| Statutory audit |
170 | 150 | ||||||
| Other assurance engagements |
0 | 0 | ||||||
| Tax advisory services |
0 | 0 | ||||||
| Other services |
105 | 142 | ||||||
|
|
|
|
|
|||||
| 275 | 292 | |||||||
|
|
|
|
|
|||||
7. FINANCIAL EXPENSES (NET)
| 2015 | 2014 | |||||||
| Interest expense |
(3,984 | ) | (2,478 | ) | ||||
| Interest income |
6 | 6 | ||||||
| Foreign exchange (net) — realized |
796 | 18 | ||||||
| Foreign exchange (net) — unrealized |
0 | (1,333 | ) | |||||
|
|
|
|
|
|||||
| Financial expenses for the period |
(3,182 | ) | (3,787 | ) | ||||
|
|
|
|
|
|||||
8. TAX EXPENSES
| 2015 | 2014 | |||||||
| Calculated income tax for the period |
0 | 0 | ||||||
| Not recognised deferred tax for the year |
(2,896 | ) | 0 | |||||
| Deferred tax for the year |
0 | (952 | ) | |||||
| Tax reimbursement for the year |
(5,875 | ) | (6,250 | ) | ||||
| Calculated tax for the year, 23,5% (2014: 24,5%) |
(8,771 | ) | (7,202 | ) | ||||
|
|
|
|
|
|||||
| Tax reimbursement for the year |
(5,875 | ) | (6,250 | ) | ||||
| Change in deferred tax |
2,516 | (952 | ) | |||||
| Tax for the year |
(3,359 | ) | (7,202 | ) | ||||
| Effective tax rate |
9.0 | % | 24.5 | % | ||||
| Tax on other comprehensive income for the period |
0 | 0 | ||||||
The long-term deferred tax asset has been evaluated against the future income within the next three fiscal years and will not be recognised in the balance sheet, as they are not realizable.
The long-term deferred tax asset amount is DKK 5.6 million as of 31 December 2015.
9. EARNINGS PER SHARE (EPS)
| 2015 | 2014 | |||||||
| Net profit/(loss) |
(33,853 | ) | (22,151 | ) | ||||
| Average number of outstanding shares* |
15,055.150 | 15,055.150 | ||||||
| Earnings per share (EPS) |
(2.25 | ) | (1.47 | ) | ||||
|
|
|
|
|
|||||
| Diluted earnings per share (DKK) including warrants (31.12.2015) |
(2.21 | ) | (1.47 | ) | ||||
|
|
|
|
|
|||||
| Diluted earnings per share (DKK) including warrants (31.03.2016) |
(0.21 | ) | (0.13 | ) | ||||
| * | Average number of outstanding shares is based on issued shares of 15,055,150 and therefore not including major shareholder loan that is converted and recognised as equity as of 31 December 2015. |
141
Table of Contents
10. ALLOCATION OF LOSS
| 2015 | 2014 | |||||||
| It is proposed that the year’s consolidated loss is transferred to retained earnings |
(33,853 | ) | (22,151 | ) | ||||
11. DEVELOPMENT PROJECTS
| 2015 | 2014 | |||||||
| Costs at the beginning of the year |
30,186 | 13,271 | ||||||
| Additions in the period |
34,776 | 16,916 | ||||||
|
|
|
|
|
|||||
| Costs end of period |
64,962 | 30,186 | ||||||
|
|
|
|
|
|||||
| Depreciation and write-down at the beginning of the year |
769 | 769 | ||||||
| Depreciation in period |
0 | 0 | ||||||
| Write-down in period |
5,430 | 0 | ||||||
|
|
|
|
|
|||||
| Depreciation and write-down end of period |
6,199 | 769 | ||||||
|
|
|
|
|
|||||
| Book value end of period |
58,763 | 29,417 | ||||||
|
|
|
|
|
|||||
All capitalised development costs are related to development work in progress.
Write-down is linked to development projects related to current products but regarded as out of scope in the current strategy for Serendex. The write-down amounts to DKK 5,4 million (2014: DKK 0 million).
In order for costs to be qualified in the balance sheet as development costs, the nature of the expense has to be linked to a specific activity in the development process. Development costs, which do not fulfil the requirements for recognition in the balance sheet, are expensed immediately in the income statement.
The development costs directly recognised in the income statement in 2015 is DKK 1.6 million for the Group (2014: DKK 4.2 million) exclusive the write-down of DKK 5,4 million.
12. TANGIBLE ASSETS
| 2015 | 2014 | |||||||
| Costs at the beginning of the year |
313 | 117 | ||||||
| Addition in period |
0 | 313 | ||||||
| Disposals in period |
0 | (117 | ) | |||||
|
|
|
|
|
|||||
| Costs end of period |
313 | 313 | ||||||
|
|
|
|
|
|||||
| Depreciation and write-down at the beginning of the year |
35 | 27 | ||||||
| Reversed depreciation on disposals |
0 | (27 | ) | |||||
| Depreciation in period |
59 | 35 | ||||||
| Write-down in period |
0 | 0 | ||||||
|
|
|
|
|
|||||
| Depreciation and write-down end of period |
94 | 35 | ||||||
|
|
|
|
|
|||||
| Book value end of period |
219 | 278 | ||||||
|
|
|
|
|
|||||
The tangible assets consists of leasehold improvements and office equipment related to Slotsmarken 17, Hørsholm.
142
Table of Contents
13. DEPOSITS
| 2015 | 2014 | |||||||
| Deposit at the beginning of the year |
194 | 134 | ||||||
| Disposals during the period |
0 | (134 | ) | |||||
| Additions during the period |
5 | 194 | ||||||
|
|
|
|
|
|||||
| Deposit end of period |
199 | 194 | ||||||
|
|
|
|
|
|||||
14. TAX RECEIVABLES AND DEFERRED TAX
| 2015 | 2014 | |||||||
| Deferred tax at the beginning of the year |
8,766 | 2,814 | ||||||
| Tax Credit Reimbursement |
(6,250 | ) | (1,250 | ) | ||||
| Deferred tax net change |
8,961 | 7,202 | ||||||
|
|
|
|
|
|||||
| Deferred tax end of period |
11,477 | 8,766 | ||||||
|
|
|
|
|
|||||
| The deferred tax concerns |
||||||||
| Intangible assets |
(12,848 | ) | (6,339 | ) | ||||
| Tangible assets |
9 | 15 | ||||||
| Loss carried forward |
24,316 | 15,090 | ||||||
|
|
|
|
|
|||||
| 11,477 | 8,766 | |||||||
|
|
|
|
|
|||||
| The deferred tax is reconciled as follows |
||||||||
| Deferred tax end of periodA |
11,477 | 8,766 | ||||||
| Long-term deferred tax not recognised in the balance sheetB |
(5,602 | ) | 0 | |||||
|
|
|
|
|
|||||
| 5,875 | 8,766 | |||||||
|
|
|
|
|
|||||
| A) | The long-term deferred tax asset has been evaluated against the future taxable income within the next three fiscal years and will not be recognized in the balance sheet due to change in accounting estimates of the future value. The long-term deferred tax asset amount is DKK 5.6 million as of 31 December 2015 (2014: 0 DKK). |
| B) | Due to tax credit reimbursement instalment by the Danish government, Serendex Group expects to be reimbursed DKK 5.875 million of the tax asset in Q4 2016. |
15. INVENTORY
| 2015 | 2014 | |||||||
| Raw materials and consumables |
197 | 283 | ||||||
| Work in progress |
343 | 714 | ||||||
| Manufactured goods and goods for resale |
704 | 251 | ||||||
|
|
|
|
|
|||||
| Total |
1,244 | 1,248 | ||||||
|
|
|
|
|
|||||
143
Table of Contents
16. LOANS FROM SHAREHOLDERS
| 2015 | 2014 | |||||||
| Loans at the beginning of the year |
23,464 | 10,025 | ||||||
| Additions during the period |
36,000 | 12,000 | ||||||
| Interest |
3,726 | 2,315 | ||||||
|
|
|
|
|
|||||
| 63,190 | 24,376 | |||||||
|
|
|
|
|
|||||
| Repayment in the period |
0 | (912 | ) | |||||
| Converted to equity |
(62,022 | ) | 0 | |||||
| Loans at the end of the period |
1,168 | 23,464 | ||||||
Agreed and signed on 10 December 2015 it was decided to convert loans of DKK 62.0 million into shares — hence the amount has been converted and recognised as equity as of 31 December 2015.
The shareholders have provided a secured undrawn committed credit facility of DKK 25 million in 2016.
17. PLEDGED ASSETS AND SECURITIES
In security for a loan to shareholder, the parent company Serendex has pledged shares in the subsidiaries of book value DKK 26.7 million (2014: 26.7 million).
18. CONTRACTUAL OBLIGATIONS AND PENDING LITIGATIONS
Obligations on rental properties
As of 31 December 2015 Serendex has total commitments of DKK 1.5 million until 2019.
Pending litigations
As of 31 December 2015 Serendex has made an external legal assessment of alleged claims against the company. None has been seen as having any material impact — hence, no provisions have been made.
19. FINANCIAL RISKS
Serendex is primarily exposed to exchange rate risks in the countries where Serendex has its main activities. I.e. the risks relate to the rise/fall in EURO, GBP, USD and NOK. It is Group policy not to actively conduct speculation in any financial risks and it is management’s strategy to seek to offset exchange-rate risks by matching revenue, as well as other positive cashflow, against costs in the same currencies.
20. RELATED-PARTY TRANSACTIONS
Related parties comprise the company’s Executive Management, Board of Directors and the major shareholder. All transactions between the related parties are based on the principle of “arm’s length”. In 2015 Serendex had the following related party transactions:
Legal services DKK 642k, from Bech-Bruun Law Firm. Partner Christian Vinding Thomsen serves as board member in Serendex.
Interest DKK 3.7 million to Sorana A/S, which is owned by board member Lorenz Jørgensen.
Remuneration paid to the members of the Executive Management and the Board of Directors. Please see note 3 for information.
144
Table of Contents
As of 15 March 2016 board member Lorenz Jørgensen and his related parties hold 85.5% of the total shares in Serendex A/S. No other shareholder holds more than 5% of the shares.
Loan and conversion of loan from shareholder (note 16).
21. STATEMENT OF CASH FLOWS — ADJUSTMENTS
| 2015 | 2014 | |||||||
| Financial income and expenses |
3,182 | 3,787 | ||||||
| Amortisation and depreciation |
5,489 | 124 | ||||||
| Adjustments in tangible assets |
0 | 227 | ||||||
| Share based incentive |
651 | 0 | ||||||
|
|
|
|
|
|||||
| Total adjustments |
9,322 | 4,138 | ||||||
|
|
|
|
|
|||||
22. STATEMENT OF CASH FLOWS — CHANGE IN WORKING CAPITAL
| 2015 | 2014 | |||||||
| Net change in receivables |
1,237 | (1,150 | ) | |||||
| Reduction in tax credit reimbursement |
(375 | ) | 0 | |||||
| Net change in inventory |
5 | 51 | ||||||
| Net change in current debt |
5,836 | 1,549 | ||||||
|
|
|
|
|
|||||
| Total change in working capital |
6,703 | 450 | ||||||
|
|
|
|
|
|||||
23. RISK OVERVIEW
Serendex is exposed to uncertainties and risk factors, which may affect some or all of the company’s activities.
Development risks
Drug development involves considerable risks. The average development period is typically more than 10 years, costs are high and the probability of reaching the market is relatively low. However, the foundation of Serendex’s business model is to produce and develop well-known biological products, which have previously been used systemically, into unique products for inhalation. Hence, the repositioning approach reduces pre-clinical and clinical risks, development costs, as well as the overall time to market.
That said, Serendex is still exposed to development risks and the following factors are assessed regularly for all development programmes:
The occurrence of unexpected and adverse side effects developed by inhalation or inducting the drug candidate into the lungs; this risk is highest in the early phases of development (preclinical and phase I) and confidence increases as the total number of patients who have been exposed to the product increases
The scientific rationale may be based on preclinical models and literature data. The early exploratory patient studies will provide an indication as to whether or not this rationale can be applied to the human setting
The complexity of clinical development, access to patients, and the speed of which an indication of a clinical effect can be established may affect the timelines of the planned clinical phase II/III development
Regulatory assessment of the drug candidate’s efficacy, safety profile and probability of final approval is not completed until phase III data are available
145
Table of Contents
Commercial risks
The flowing factors are assessed in connection with the initiation of a drug development programme and are evaluated in connection with reassessing the pipeline:
Degree and scope of patent protection
Market size (prevalence and expected growth in patients)
Competitive situation (existing treatment as well as new drugs under development with the same scientific rationale)
Development time and associated costs
Interest from potential partners
Market access
Contractual risks
Serendex’s business model is founded upon an extensive outsourcing strategy and international strategic alliances. Thus it is essential to secure that vendor contracts or other agreements do not impose abnormal obligations on Serendex, nor are drafted in an unbalanced manner with regards to the protection of Serendex’s business. Therefore, before entering any agreements, partners are thoroughly evaluated with regards to financial solidity, delivery quality, timeliness as well as overall reliability.
Employee risks
Serendex is well aware that employees are an important asset. As Serendex’s business model is founded upon an extensive outsourcing strategy, having the right competencies with the adequate experience is vital. Therefore, it is important that Serendex continues to attract, retain and develop skilled employees. Failure to do so will negatively impact the Company’s continued development.
Financial risks
Serendex is primarily exposed to interest risks in connection with surplus liquidity and interest-bearing liabilities as the non-current loans are stablished at a fixed interest rate. Interest is added to surplus liquidity in accordance with the development of the day to day interest in Danske Bank between 0-1%.
Further, Serendex is primarily exposed to exchange rate risks in the countries where Serendex has its main activities. I.e. the risks relate to the rise/(fall) in the British pound, American dollar, Norwegian kroner and Euro. As of 31 December 2015, a realized currency gain of DKK 0.8 million was recognised as a financial income, which primarily was related to the increase in the Norwegian kroner compared to the Danish kroner in the beginning of 2015. At the end of 2015 a total increase in Norwegian kroner vs. Danish kroner of 10 % will result in a decrease in operating profit of DKK 0.0 million (2014: DKK 1.8 million). A total increase in British pounds vs. Danish kroner of 10 % will result in a decrease in operating profit of DKK 0.0 million (2014: DKK 0.2 million). A total increase in American dollars vs. Danish kroner of 10 % will result in a decrease in operating profit of DKK 0.1 million (2014: DKK 0.7 million). A total increase in Euro vs. Danish kroner of 10 % will result in a decrease in operating profit of DKK 0.1 million (2014: DKK 0.0 million). It is Group policy not to
146
Table of Contents
actively conduct speculation in financial risks and it is management’s strategy to seek to offset exchange-rate risks by matching revenue and costs in the same currencies.
| LIQUIDITY RISK AS OF 31 December 2015 |
31-12-2015 | 0-1 year | 1-5 years | |||||||||
| Loan, non-current |
1,168 | 1,555 | ||||||||||
| Lease liability |
1,700 | 425 | 1,275 | |||||||||
| Trade payables |
5,921 | 5,921 | ||||||||||
| Other current liabilities |
5,108 | 5,108 | ||||||||||
|
|
|
|
|
|
|
|||||||
| Total liabilities |
13,897 | 11,454 | 2,830 | |||||||||
|
|
|
|
|
|
|
|||||||
| Cash |
5,974 | — | ||||||||||
| Receivables, non-current |
— | — | ||||||||||
| Receivables |
6,992 | 6,992 | ||||||||||
| Other expenses |
— | — | ||||||||||
|
|
|
|
|
|||||||||
| Financial assets |
12,966 | 6,992 | ||||||||||
|
|
|
|
|
|||||||||
| Liquidity risk |
931 | 4,463 | 2,830 | |||||||||
Serendex has secured an undrawn committed credit facility provided by the major shareholder, which provides Serendex with a satisfactory cash situation for 2016.
Capital resources
Serendex intends to license its products to pharmaceutical companies and thereby derive income from a combination of fixed payments and ongoing royalty income. Until Serendex has established such a license agreement, Serendex will be a capital-consuming company due to investments in drug development and in further strengthening of the pipeline. Therefore, it is vital that the Company always has sufficient financial resources. The Board of Directors receives reports on a monthly basis, which include information dealing with the amount and scope of Serendex’s financial resources. Moreover, at each board meeting, the financial resources are assessed in regards to the potential of procuring necessary capital.
Serendex has a satisfactory cash situation for 2016 to continue the phase IIb clinical trial of GM-CSF for ARDS and the pivotal phase II/III clinical trial of Molgradex® for PAP according to plans. Hence, the annual report for 2015 has been prepared for on-going business. In order to pursue the development strategy as outlaid in the Management Report, Serendex is dependent on acquiring additional capital to continue operations 2017 onwards. Serendex will therefore continue to investigate opportunities and terms for entering into strategic partnerships or mergers and/or licensing agreements that will strengthen Serendex’s financial position. In addition, the company will investigate opportunities for receiving additional debt or equity financing.
Securing the company’s operation and assets
Serendex has taken out insurance to cover both any losses due to claims in connection with clinical studies and the named patient sales programme as well as the loss of assets due to fire, water damage, theft, and so forth. All insurance related issues are handled by an external insurance broker who reports yearly as to whether the company’s insurance cover is sufficient and reasonable.
24. SIGNIFICANT EVENTS OCCURRING AFTER THE BALANCE SHEET DATE
The rights issue initiated in 2015 was fully subscribed when the subscription period ended 4 January 2016.
Serendex has in 2016 secured an undrawn committed credit facility provided by the major shareholder that supports a satisfactory cash situation for Serendex for 2016.
147
Table of Contents
Oslo Stock Exchange has in 2016 approved the delisting of Serendex from Oslo Axess. This means that Serendex as of 4 May 2016 no longer is a listed company
As of 15 July 2016 Serendex Pharmaceuticals A/S has signed a business agreement with Savara Inc, Texas (USA), including transfer of the entire operation and all activities.
Serendex Pharmaceuticals A/S has
| • | changed its chairman of the board as of 16 July 2016. |
| • | changed its CEO as of 16 July 2016. |
| • | changed its name to Serenova A/S as of 5 September 2016. |
| • | changed its business address as of 2 September 2016. |
No other significant events have occurred subsequent to the balance sheet date that are considered to have a material influence in the evaluation of the 2015 report.
25. BOARD OF DIRECTORS AND EXECUTIVE MANAGEMENT
BOARD OF DIRECTORS
| Karin Verland |
Other positions | |
| Chairman |
Nygart Privathospital A/S (chairman), | |
| Board member since 2014 |
N2MO (board member) and Justitia (board member). |
As of 01.03.2016 Karin Verland and her related parties hold 234,110 Serendex shares.
| Lorenz Johannes Thorndahl Jørgensen |
Other positions | |
| Board member since 2013 Sorana A/S (board member), LJ investering ApS (board |
||
| Member of Audit committee member), Investeringsselskabet Fir A/S International (board member), Fagus A/S (board member), Scanpol International ApS (board member), Triton Hotel A/S (board member), Musholmfonden (board member), Sorø Kunstmuseum (board member). | ||
As of 01.03.2016 Lorenz Jørgensen and his related parties hold 140,369,193 Serendex shares.
| Christian Vinding Thomsen |
Other positions | |
| Board member since 2013 |
||
| Bech-Bruun Law firm (partner), KT Stålindustri A/S (chairman), RAC Denmark A/S (AVIS & Budget) (board member) and Mark & Gerstenberg A/S (board member). | ||
As of 01.03.2016 Christian Vinding Thomsen and his related parties hold 50,000 Serendex shares.
| Søren Bech Justesen |
Other positions | |
| Board member since 2016 |
Conscia Holdning A/S (CFO and member of executive | |
| Member of Audit committee |
management), AX IV CON II ApS (member of Executive management) and Conscia A/S (board member). |
As of 01.03.2016 Søren Bech Justesen and his related parties hold 100,000 Serendex shares and 80,000 warrants.
Don deBethizy, Helena Nordin Rudberg and Tone Bjørnov resigned from the Board of Directors with effect from the extraordinary general meeting of 9 February 2016.
148
Table of Contents
EXECUTIVE MANAGEMENT
Kim Arvid Nielsen
CEO and member of Executive Management since 2013
As of 01.03.2016 Kim Arvid Nielsen and his related parties hold 114,746 Serendex shares and 120,000 warrants.
Søren Bech Justesen resigned from Executive Management January 2016
26. ACCOUNTING POLICIES
Accounting policies applied in the preparation of the consolidated financial statements are set out below. The accounting policies are unchanged compared to 2014.
New standards and interpretations
Based on an assessment of new or amended and revised accounting standards and interpretations (‘IFRSs’) issued by the International Accounting Standards Board (IASB) effective on or after 1 January 2015, it has been assessed that the application of these new IFRSs has not had a material impact on the Consolidated financial statements in 2015, and Management does not anticipate any significant impact on future periods from the adoption of these new IFRSs.
IASB has issued a number of new or amended and revised accounting standards and interpretations that have not yet come into effect. In general, the following standards are expected to have the most significant impact on current accounting regulation:
| • | IASB has issued IFRS 9 ‘Financial Instruments’, with effective date 1 January 2018. The Management does currently not see this amendment relevant for Serendex Pharmaceuticals A/S. |
| • | IASB has issued IFRS 15 ‘Revenue from contracts with customers’, with effective date 1 January 2018. The Management does currently not see this amendment relevant for Serendex Pharmaceuticals A/S. |
| • | IASB has issued IFRS 16 ‘Leases’ with effective date 1 January 2019. The Management does currently not see this amendment relevant for Serendex Pharmaceuticals A/S. |
Basis of preparation
The Annual Report has been prepared in accordance with the International Financial Reporting Standards (IFRS) as issued by the IASB.
The preparation of financial statements in conformity with IFRS requires the use of certain critical accounting estimates. It also requires management to exercise its judgement in the process of applying the Serendex Pharmaceuticals A/S Group’s accounting policies. The areas involving a higher degree of judgment or complexity, and areas where assumptions and estimates are significant to the consolidated financial statements are disclosed.
Serendex has a satisfactory cash situation for 2016. Thus, the annual report for 2015 has been prepared for on-going business.
The consolidated financial statements are presented in DKK, reflecting the company’s functional currency.
149
Table of Contents
Basis of consolidation
The consolidated financial statements are prepared by adding the audited financial statements of the parent company and the individual subsidiaries, all of which are prepared in accordance with the group’s accounting policies.
The following companies are consolidated:
| • | Serendex Pharmaceuticals A/S (parent company) |
| • | Drugrecure ApS (100% Serendex) |
| • | Pharmaorigin ApS (100% Serendex) |
Recognition and measurement in general
The net revenue is recognised in the profit and loss account if delivery and risk transfer to the buyer have taken place before the end of the year, and if the income can be determined reliably and is expected to be received. The net revenue is recognised exclusive of VAT and taxes and with the deduction of any discounts granted in connection with the sale.
Recognition of value adjustments of assets and liabilities are recognised in the profit and loss account upon financial assessment.
All costs — including depreciation, amortisation, write-down, provisions, and reversals, which are due to changes in estimated amounts previously recognised in the profit and loss account — are recognised in the profit and loss account.
Assets are recognised in the balance sheet when the company is liable to achieve future, financial benefits and the value of the asset can be measured reliably.
Liabilities are recognised in the balance sheet when the company is liable to lose future, financial benefits and the value of the liability can be measured reliably.
Translation of foreign currency
Operational transactions in foreign currency are translated by using the exchange rate at cost basis upon bank transaction. Differences in the rate of exchange arising between the rate at the date of transaction and the rate at the date of payment are recognised in the profit and loss account as an item under net financials.
Debtors, creditors, and other monetary items in foreign currency — not settled at the date of the balance sheet — are translated by using the period closing rate held by The Danish Central Bank. The difference between the closing rate and the rate at the time of establishment of the receivable or the payable is recognised in the profit and loss account under financial income and financial costs.
Fixed assets and other non-monetary assets acquired in foreign currency and which are not considered to be investment assets purchased in foreign currencies are measured at the exchange rate on the transaction date.
INCOME STATEMENT
Net revenue
As of 31 December 2015 Serendex has only one segment according to IFRS.
Revenue represents amounts receivable for products or services delivered in the normal course of business of the company. Revenue is reduced for estimated customer returns and other similar allowances whenever applicable
150
Table of Contents
based on historical data and expectations of future sales. Revenue is recognised upon invoiced sale and when risks and rewards of ownership is transferred to the customer. The risks and rewards of ownership are generally transferred at the time the product is shipped and delivered to the customer. Revenue is recognised in the profit and loss account when management has established that all aforementioned conditions for revenue recognition have been met.
Other operating income and costs comprise accounting items of secondary nature in proportion to the principal activities of the enterprise.
Up-front payments that are attributable to subsequent research and development activities are recognised as deferred revenue and will subsequently be recognised as revenue over the expected contract period. Non-refundable up-front payments and milestone payments that are not attributable to subsequent research and/or development activities or other delivery obligations are recognised as revenue when the contracts are signed or when the milestone criteria are met respectively.
Cost of goods sold
The cost of goods sold comprises costs paid for manufacturing in order to generate net revenue for the year including depreciation, amortisation and write-downs of inventory.
Staff expenses
Staff expenses comprise total remuneration to Serendex employees including fees to Board of Directors.
Raw materials and consumables used
Raw materials and consumables used comprise handling charges, distributions costs and costs paid for manufacturing samples and references.
External expenses
External expenses compromise all external costs including development costs, which are not directly attributable to the Company’s development of new products (capitalised costs). External expenses includes depreciation and write-downs.
The classification of costs (income statement vs. equity), associated with the rights issue is in accordance with IAS32. I.e. costs directly attributable to issuing shares or expected Addition of equity are deducted from equity and costs related to the stock market listing, or otherwise not incremental and directly attributable to issuing new shares, are recognised as an external expense in the income statement.
NET FINANCIALS
Net financials include interest income, interest expenses on loans, and realized and unrealized exchange rate gains and losses. Net financials are recognised in the profit and loss account with the amounts concerning the financial year.
Tax
Tax comprises the current tax for the year and the changes in deferred tax. Tax costs are recognised in the profit and loss account with the amounts concerning the fiscal year with the share referring to entries in the equity subsequently deferred tax asset.
151
Table of Contents
BALANCE SHEET ITEMS
Intangible assets
Intangible fixed assets comprise development projects, patents, and licenses. Development costs comprise costs directly and indirectly attributable to development of new products from which the Company expects a future economic benefit.
All other development costs are recognised as costs in the profit and loss accounts.
Capitalised development costs are measured at cost with deduction of accrued amortisations or at the recoverable value, if this is lower.
The carrying amounts of intangible assets carried at cost or amortized cost are tested annually to determine whether there are indications of any impairment in excess of that expressed in normal amortisation. If that is the case, the asset is written down to the recoverable amount, which is the higher value of the net sales price and the capitalised value. Impairment losses on intangible assets are recognised under the same line item as amortisation of the assets.
For development projects in progress, the recoverable amount is assessed annually, regardless of whether any indications of impairment have been found.
After completion of the development work, capitalised development costs are amortized on a straight line basis over the estimated financial useful life.
Profit and loss from the realization of development projects, patents, and licenses are measured as the difference between the sales price with deduction of sales costs and the book value at the time of the sale.
Tangible assets
Tangible assets are measured at cost with deduction of accrued depreciation and write-down.
The basis of depreciation is costs with deduction of expected residual value after the end of the useful life of the asset.
The cost comprises the Addition cost and costs directly attached to the Addition until the time when the asset is ready for use.
Depreciation takes place on a straight-line basis and based on an evaluation of the expected useful life:
Office equipment and fittings: 3 years
IT and software licenses: 2 years
Leasehold improvements: 10 years
Minor assets with an expected useful life of less than 1 year and/or of a cost less than EUR 2,000 (app. DKK 15,000) are recognised as costs in the profit and loss account in the year of Addition.
Profit or loss deriving from the sales of tangible fixed assets is measured as the difference between the sales price reduced by the selling costs and the book value at the time of the sale. Profit or loss is recognised in the profit and loss account under depreciation.
152
Table of Contents
Write-down of assets
The book values of intangible as well as tangible fixed assets are subject to annual write-down assessment in order to disclose any indications of impairment beyond those expressed by amortisation and depreciation respectively.
If indications of impairment are disclosed, impairment tests are carried out for each individual asset or group of assets respectively. Write-down takes place to the recoverable amount, if this value is lower than the book value.
The recoverable value is equal to the value of the net selling price or the value in use, whichever is higher. The value in use is determined as the present value of the expected net income deriving from the use of the asset or the group of assets. Any loss based on the write-down test is recognised in the profit and loss account under depreciation.
Inventories
Inventories are measured at cost on basis of measured average prices. In case the net realizable value is lower than the cost, write-down takes place at this lower value.
The inventory includes:
Addition of pharmaceutical ingredients, which include the cost for raw materials and the initial processing
The cost for manufactured goods and works in progress
The net realizable value for inventories is recognised as the market price with the deduction of completion costs and selling costs, and it is determined by taking negotiability, obsolescence, and the development of the expected market price into consideration. All logistic costs related to the inventories are recognised in the profit and loss account.
Deferred tax
Long-term deferred tax (+12 months) and current tax (less than 12 months) are recognised in the balance sheet at the amount calculated on the basis of the expected taxable income for the year adjusted for tax on previous years, taxable income and prepaid taxes. Tax receivable and tax liabilities are set off to the extent that legal right of set-off exists and if the items are expected to be settled net or simultaneously.
Deferred tax is measured on the basis of all temporary differences in assets and liabilities with a balance sheet focus.
Deferred tax is measured based on the tax rules and tax rates applying under the legislation on the balance sheet date and prevailing when the deferred tax is expected to be released as current tax. In the period 2014 to 2016, the corporate tax rate will be reduced gradually from 25% to 22%, which will affect the deferred tax liabilities and deferred tax assets. Unless a recognition with a different tax rate than 22% will result in a significant material deviation in the estimated deferred tax liability or tax asset, deferred tax liabilities and assets are recognised by 22%.
Trade receivables
Trade receivables are recognised at amortized cost less potential losses on doubtful debts. Write-downs are based on individual assessments of each debtor.
153
Table of Contents
Other receivables, prepayments and accrued expenses
Deposits comprise rental deposits paid to real estate agencies.
Prepaid expenses paid in advance but which has not yet been incurred are recognised under assets.
Accrued expenses recognised under assets comprise incurred costs concerning the next financial year.
Cash and cash equivalents
Cash and cash equivalents includes cash in Danske Bank.
Non-current liabilities
Non-current liabilities comprise long term loans to management and corresponds to the outstanding debt of the loan.
Current liabilities
Current liabilities are measured at amortized costs, which usually corresponds to the nominal value.
CASH FLOW STATEMENT
The cash flow statement shows the cash flow of the company for the year, divided in cash flows deriving from
Operating activities
Capitalised activities
Financing activities
Changes in the liabilities
Available funds at the beginning and the end of the year respectively
Cash flow from operating activities
Cash flow from operating activities is calculated as the profit and loss results for the year adjusted for non-cash operating items, the change in the working capital, and corporate tax paid/received.
Cash flow from capitalised activities
Cash flow from investment activities comprises development costs directly attributable to the Company’s research and development of new products and payments in connection with the Addition tangible assets.
Cash flow from financing activities
Cash flow from financing activities comprises changes in the size or the composition of the share capital and the costs in this connection. Furthermore, these activities comprise borrowings, instalments on interest bearing debt, and payment of dividend to the shareholders.
27. STATEMENT OF THE BOARD OF DIRECTORS
The Board of Directors have on February 7, 2017 approved the consolidated financial statements of Serendex Pharmaceuticals A/S for the period 1 January 2015 to 31 December 2015.
154
Table of Contents
Unaudited Consolidated Income Statement
for the period 1 January — 30 June
| DKK thousand |
Notes | half year 2016 |
half year 2015 |
|||||||||
| Net revenue |
704 | 147 | ||||||||||
| Cost of goods sold |
(232 | ) | (115 | ) | ||||||||
|
|
|
|
|
|||||||||
| Gross profit |
471 | 32 | ||||||||||
|
|
|
|
|
|||||||||
| Staff expenses |
3,4 | (8,698 | ) | (9,256 | ) | |||||||
| External expenses |
4 | (4,633 | ) | (4,106 | ) | |||||||
| Other expenses |
4 | (5,424 | ) | (520 | ) | |||||||
|
|
|
|
|
|||||||||
| Operating profit/loss (-) |
(18,284 | ) | (13,851 | ) | ||||||||
|
|
|
|
|
|||||||||
| Net financials |
(328 | ) | (561 | ) | ||||||||
|
|
|
|
|
|||||||||
| Profit/loss (-) before tax |
(18,612 | ) | (14,412 | ) | ||||||||
|
|
|
|
|
|||||||||
| Tax expenses |
0 | 3,170 | ||||||||||
|
|
|
|
|
|||||||||
| Net profit/loss (-) |
(18,612 | ) | (11,242 | ) | ||||||||
|
|
|
|
|
|||||||||
STATEMENT OF COMPREHENSIVE INCOME
| DKK thousand |
||||||||
| Net profit/loss (-) |
(18,612 | ) | (11,242 | ) | ||||
| Other comprehensive income |
0 | 0 | ||||||
|
|
|
|
|
|||||
| Total comprehensive income |
(18,612 | ) | (11,242 | ) | ||||
|
|
|
|
|
|||||
155
Table of Contents
Unaudited Consolidated Balance Sheet
| ASSETS | ||||||||||||
| DKK thousand |
Notes | half year 2016 |
December 31, 2015 |
|||||||||
| NON-CURRENT ASSETS |
||||||||||||
| Intangible assets |
||||||||||||
| Development projects |
5 | 76,023 | 58,763 | |||||||||
| Tangible assets |
||||||||||||
| Plant and equipment |
6 | 191 | 219 | |||||||||
| Financial assets |
||||||||||||
| Long-term deferred tax |
0 | 0 | ||||||||||
| Non-current receivables |
||||||||||||
| Deposits |
199 | 199 | ||||||||||
|
|
|
|
|
|||||||||
| Total non-current assets |
76,412 | 59,182 | ||||||||||
|
|
|
|
|
|||||||||
| CURRENT ASSETS |
||||||||||||
| Inventories |
118 | 1,244 | ||||||||||
| Receivables |
||||||||||||
| Tax receivables |
7 | 5,875 | 5,875 | |||||||||
| Trade receivables |
0 | 0 | ||||||||||
| Other receivables |
562 | 1,117 | ||||||||||
|
|
|
|
|
|||||||||
| Total receivables |
6,437 | 6,992 | ||||||||||
|
|
|
|
|
|||||||||
| Cash and cash equivalents |
2,398 | 5,914 | ||||||||||
| Total current assets |
8,849 | 14,210 | ||||||||||
|
|
|
|
|
|||||||||
| Total assets |
85,366 | 73,391 | ||||||||||
|
|
|
|
|
|||||||||
| EQUITY AND LIABILITIES | ||||||||||||
| EQUITY |
||||||||||||
| Share capital |
16,410 | 12,134 | ||||||||||
| Retained earnings |
52,046 | 49,059 | ||||||||||
|
|
|
|
|
|||||||||
| Total equity |
68,456 | 61,193 | ||||||||||
|
|
|
|
|
|||||||||
| LIABILITIES |
||||||||||||
| Non-current liabilities |
||||||||||||
| Long-term loans from shareholders and management |
8 | 11,168 | 1,168 | |||||||||
|
|
|
|
|
|||||||||
| Total non-current liabilities |
11,168 | 1,168 | ||||||||||
|
|
|
|
|
|||||||||
| Current liabilities |
||||||||||||
| Trade payables |
3,150 | 5,921 | ||||||||||
| Other current liabilities |
2,592 | 5,108 | ||||||||||
|
|
|
|
|
|||||||||
| Total current liabilities |
5,742 | 11,029 | ||||||||||
|
|
|
|
|
|||||||||
| Total liabilities |
16,910 | 12,197 | ||||||||||
|
|
|
|
|
|||||||||
| Total equity and liabilities |
85,366 | 73,391 | ||||||||||
|
|
|
|
|
|||||||||
156
Table of Contents
Unaudited Consolidated Changes in equity
| DKK thousand |
Notes | Share capital | Retained earnings |
Total | ||||||||||||
| Equity at 1 January 2015 |
1,506 | 32,179 | 33,685 | |||||||||||||
| Profit/loss (-) |
0 | (33,853 | ) | (33,853 | ) | |||||||||||
| Other comprehensive income |
0 | 0 | 0 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total comprehensive income |
0 | (33,853 | ) | (33,853 | ) | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Share-based incentive |
3 | 0 | 651 | 651 | ||||||||||||
| Share capital increase |
10,628 | 0 | 10,628 | |||||||||||||
| Share premium by converted shareholder loan |
0 | 51,394 | 51,394 | |||||||||||||
| Capital transactions costs |
0 | (1,311 | ) | (1,311 | ) | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Equity at 31 December 2015 |
12,134 | 49,059 | 61,193 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Equity at 1 January 2016 |
12,134 | 49,059 | 61,193 | |||||||||||||
| Profit/loss (-) |
0 | (18,612 | ) | (18,612 | ) | |||||||||||
| Other comprehensive income |
0 | 0 | 0 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
| Total comprehensive income |
0 | (18,612 | ) | (18,612 | ) | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Share-based incentive |
3 | 0 | 2,605 | 2,605 | ||||||||||||
| Share capital increase |
4,276 | 0 | 4,276 | |||||||||||||
| Share premium |
0 | 20,600 | 20,600 | |||||||||||||
| Capital transactions costs |
0 | (1,606 | ) | (1,606 | ) | |||||||||||
|
|
|
|
|
|
|
|||||||||||
| Equity at 30 June 2016 |
16,410 | 52,046 | 68,456 | |||||||||||||
|
|
|
|
|
|
|
|||||||||||
The share capital comprises of 16,410,113.50 shares, each with a nominal value of DKK 0.10. Shareholder loan conversion recognized end of 2015 comprises of 106,279,592 shares, also each with a nominal value of DKK 0.10. No shares hold particular rights. Conversion registered in “Erhvervsstyrelsen” on the 13.01.2016.
157
Table of Contents
Unaudited Consolidated Cash Flow Statement
for the period 1 January 2016 — 30 June
| DKK thousand |
Notes | half year 2016 |
half year 2015 |
|||||||||
| Profit/loss (-) before tax |
(18,612 | ) | (14,412 | ) | ||||||||
| Adjustments |
10 | 2,962 | 1,897 | |||||||||
| Change in working capital |
11 | (3,606 | ) | 364 | ||||||||
| Cash flow from operating activities before net financials |
(19,257 | ) | (12,151 | ) | ||||||||
| Currency gain/loss |
(146 | ) | 822 | |||||||||
| Net interest costs |
(182 | ) | (1,383 | ) | ||||||||
| Cash flow from ordinary activities |
(19,585 | ) | (12,711 | ) | ||||||||
| Tax reimbursement |
0 | 0 | ||||||||||
|
|
|
|
|
|||||||||
| Cash flow from operating activities |
(19,585 | ) | (12,711 | ) | ||||||||
|
|
|
|
|
|||||||||
| Addition of intangible assets |
5 | (17,260 | ) | (15,941 | ) | |||||||
| Addition of tangible assets |
6 | 0 | 0 | |||||||||
| Paid deposits |
0 | 14 | ||||||||||
|
|
|
|
|
|||||||||
| Cash flow from investment activities |
(17,260 | ) | (15,927 | ) | ||||||||
|
|
|
|
|
|||||||||
| Loans received from shareholders |
8 | 10,000 | 12,000 | |||||||||
| Share capital increase |
0 | 0 | ||||||||||
| Share premium by IPO |
24,876 | 0 | ||||||||||
| Capital transaction costs |
(1,606 | ) | (2,251 | ) | ||||||||
|
|
|
|
|
|||||||||
| Cash flow from financial activities |
33,270 | 9,749 | ||||||||||
|
|
|
|
|
|||||||||
| Cash flow in total |
(3,576 | ) | (18,889 | ) | ||||||||
| Cash and cash equivalents at the beginning of the year |
5,974 | 20,460 | ||||||||||
|
|
|
|
|
|||||||||
| Cash and cash equivalents end of period |
2,398 | 1,571 | ||||||||||
|
|
|
|
|
|||||||||
158
Table of Contents
DKK thousand
1. CAPITAL RESOURCES
As of 15 July 2016 Serendex A/S has signed a business agreement with Savara Inc, Texas (USA), including transfer of the entire operation and all activities. This is done in order to strengthening the financial and strategic platform for the further development of the pipeline.
Savara Inc will continue to provide the necessary funding of the Danish entities including Savara ApS, Drugrecure Aps and Pharmorigin ApS — in order to secure the continued operation of the entities going forward.
2. ESTIMATES AND JUDGEMENTS
The preparation of the consolidated financial statements requires the making of estimates and judgments that affect the reporting of assets, liabilities and expenses. The estimates and judgments are reviewed on an ongoing basis. Estimates and judgments are based on historical results and on various other assumptions, which Serendex believes to be reasonable under the circumstances. We believe that the accounting policies relating to development costs and deferred tax involve estimates or judgments by management that could have a materially affect to the reported financial position and results.
Significant accounting policies
The interim financial statements are prepared in accordance with IAS 34, Interim Financial Reporting, as issued by IASB. The interim financial statements are presented in Danish Kroner (DKK), which is considered the primary currency of the Group’s activities and the functional currency of the parent company. The accounting policies used in the interim financial statements are consistent with those used in the consolidated financial statements for 2015 and in accordance with the recognition and measurement policies in the International Financial Reporting Standards (IFRS) as issued by the ASB.
Significant accounting estimates, assumptions and uncertainties
In the preparation of the interim financial statements according to IAS 34, Interim Financial Reporting, as issued by the IASB, Management is required to make certain estimates as many financial statement items cannot be reliably measured, but must be estimated. Such estimates comprise judgments made on the basis of the most recent information available at the reporting date. It may be necessary to change previous estimates as a result of changes to the assumptions on which the estimates were based or due to supplementary information, additional experience or subsequent events. Similarly, the value of assets and liabilities often depends on future events that are somewhat uncertain. In that connection, it is necessary to set out e.g. a course of events that reflects Management’s assessment of the most probable course of events.
Further to the significant accounting estimates, assumptions and uncertainties, which are stated in the Annual Report 2015, the Management has not changed significant estimates and judgments regarding recognition and measurement.
Basis of consolidation
The interim consolidated financial statements are prepared by adding the interim financial statements of the parent company and the individual subsidiaries, all of which are prepared in accordance with the group’s accounting policies.
159
Table of Contents
The following companies are consolidated:
| • | Serendex A/S |
| • | Drugrecure ApS |
| • | Pharmaorigin ApS |
Development costs
The management is confident of achieving approval of the pipeline products, as the products are based on an existing approved drug, and hold the evidence to support this. The entire capitalized cost base of development costs is part of the signed business transfer agreement with Savara Inc.
Deferred tax
Due to tax credit reimbursement instalment by the Danish government, Serendex expects to be reimbursed DKK 5.9 million in Q4 2016 (relating to tax loss 2015). Hence the tax receivable has been recognized in the balance sheet. Due to the loss in 2016, Serendex expects to be correspondingly reimbursed in Q4 2017 (relating to tax loss in 2016), however it is not possible to estimate the amount reliable and a tax receivable relating to this reimbursement has not been recognized in the balance sheet. Long-term deferred tax asset has been evaluated against the future income within the next three fiscal years and will not be recognised in the balance sheet, as they are not realizable. The long-term deferred tax asset amounts to DKK 9.1 million as of 30 June 2016.
3. STAFF EXPENSES
| DKK thousand |
half year 2016 |
half year 2015 |
||||||
| Staff |
||||||||
| Salaries, cash bonus, etc. |
4,300 | 3,991 | ||||||
| Pension costs or other social security costs |
618 | 917 | ||||||
| Share based incentive |
1,157 | 0 | ||||||
| Other staff costs |
195 | 207 | ||||||
|
|
|
|
|
|||||
| 6,270 | 5,115 | |||||||
|
|
|
|
|
|||||
| Management |
||||||||
| Fees to Board of Directors |
292 | 717 | ||||||
| Salary, cash bonus, etc. to Executive Management |
555 | 3,072 | ||||||
| Pension contributions to Executive Management |
133 | 213 | ||||||
| Share-based incentive |
1,448 | 139 | ||||||
|
|
|
|
|
|||||
| 2,428 | 4,141 | |||||||
|
|
|
|
|
|||||
| Total staff expenses |
8,698 | 9,256 | ||||||
|
|
|
|
|
|||||
| Average number of full time employees (FTE) |
12.6 | 11.5 | ||||||
| FTEs as of end of period |
11.3 | 11.8 | ||||||
160
Table of Contents
4. OTHER EXPENSES AND COSTS BY FUNCTION
| DKK thousand |
half year 2016 |
half year 2015 |
||||||
| Other expenses |
||||||||
| Capital cost (funding) |
5,424 | 520 | ||||||
|
|
|
|
|
|||||
| 5,424 | 520 | |||||||
|
|
|
|
|
|||||
| Costs by function |
||||||||
| Sales and distribution costs |
917 | 415 | ||||||
| Expensed development costs |
1,449 | 378 | ||||||
| Staff and Administrative expenses |
10,965 | 12,570 | ||||||
|
|
|
|
|
|||||
| 18,755 | 13,883 | |||||||
|
|
|
|
|
|||||
5. DEVELOPMENT PROJECTS
| DKK thousand |
half year 2016 |
half year 2015 |
||||||
| Costs at the beginning of the year |
64,962 | 30,186 | ||||||
| Additions in the period |
17,260 | 15,941 | ||||||
|
|
|
|
|
|||||
| Costs end of period |
82,222 | 46,127 | ||||||
|
|
|
|
|
|||||
| Depreciation and write-down at the beginning of the year |
6,199 | 769 | ||||||
| Depreciation in period |
0 | 0 | ||||||
| Write-down in period |
0 | 0 | ||||||
|
|
|
|
|
|||||
| Depreciation and write-down end of period |
6,199 | 769 | ||||||
|
|
|
|
|
|||||
| Book value end of period |
76,023 | 45,357 | ||||||
|
|
|
|
|
|||||
All capitalised development costs are related to development work in progress. In order for costs to be qualified in the balance sheet as development costs, the nature of the expense has to be linked to a specific activity in the development process. Development costs, which do not fulfil the requirements for recognition in the balance sheet, are expensed immediately in the income statement. The development costs recognised in the income statement as of 30 June 2016 is DKK 2.0 million for the Group.
6. TANGIBLE ASSETS
| Costs at the beginning of the year |
313 | 313 | ||||||
| Addition in period |
0 | 0 | ||||||
| Disposals in period |
0 | 0 | ||||||
|
|
|
|
|
|||||
| Costs end of period |
313 | 313 | ||||||
|
|
|
|
|
|||||
| Depreciation and write-down at the beginning of the year |
93 | 35 | ||||||
| Reversed depreciation on disposals |
0 | 0 | ||||||
| Depreciation in period |
29 | 29 | ||||||
| Write-down in period |
0 | 0 | ||||||
|
|
|
|
|
|||||
| Depreciation and write-down end of period |
122 | 64 | ||||||
|
|
|
|
|
|||||
| Book value end of period |
191 | 249 | ||||||
|
|
|
|
|
The tangible assets consist of leasehold improvements and office equipment related to Slotsmarken 17, Hørsholm.
161
Table of Contents
7. TAX RECEIVABLES AND DEFERRED TAX
| DKK thousand |
half year 2016 |
half year 2015 |
||||||
| Deferred tax at the beginning of the year |
11,477 | 8,766 | ||||||
| Tax Credit Reimbursement 2015 |
(5,875 | ) | 0 | |||||
| Tax Credit Reimbursement as of 30 June 2016 |
(2,979 | ) | 0 | |||||
| Deferred tax net change |
6,448 | 3,170 | ||||||
|
|
|
|
|
|||||
| Deferred tax end of period |
9,071 | 11,936 | ||||||
|
|
|
|
|
|||||
| The deferred tax concerns |
||||||||
| Intangible assets |
(16,966 | ) | (9,846 | ) | ||||
| Tangible assets |
9 | 12 | ||||||
| Loss carried forward |
26,028 | 21,770 | ||||||
|
|
|
|
|
|||||
| Deferred tax end of period |
9,071 | 11,936 | ||||||
|
|
|
|
|
|||||
| The deferred tax is reconciled as follows |
||||||||
| Deferred tax end of period |
14,946 | 11,936 | ||||||
| Long-term deferred tax |
0 | 5,686 | ||||||
| Long-term deferred tax not recognised in the balance sheet |
9,071 | 0 | ||||||
|
|
|
|
|
|||||
| Current asset |
5,875 | 6,250 | ||||||
|
|
|
|
|
|||||
The long-term deferred tax asset has been evaluated against the future taxable income within the next three fiscal years and will not be recognised in the balance sheet, as they are not realizable. Due to tax credit reimbursement instalment by the Danish government, Serendex Group was cash reimbursed DKK 5.875 million of the tax asset in November 2016.
8. LOANS FROM SHAREHOLDERS
| Loans at the beginning of the year |
1,168 | 24,448 | ||||||
| Additions during the period |
10,000 | 12,000 | ||||||
|
|
|
|
|
|||||
| Loans at the end of the period |
11,168 | 36,598 | ||||||
|
|
|
|
|
|||||
| Interest (accrued) |
181 | 0 | ||||||
| Loans including unpaid interest at the end of the period |
11,349 | 36,598 | ||||||
|
|
|
|
|
The shareholders have provided a secured and committed credit facility of DKK 25 million of which DKK 15.0 million is undrawn as of 30 June 2016.
9. FINANCIAL RISKS
Serendex is exposed to exchange rate risks in the countries where Serendex conducts its business i.e. the risk relates to the rise/fall in EURO, GBP and USD. It is Group policy not to actively conduct speculation in any financial risks and it is the management’s strategy to seek to offset exchange-rate risks.
162
Table of Contents
10. STATEMENT OF CASH FLOWS — ADJUSTMENTS
| DKK thousand |
Unaudited Notes | half year 2016 |
half year 2015 |
|||||||||
| Financial income and expenses |
328 | 561 | ||||||||||
| Amortisation and depreciation |
28 | 29 | ||||||||||
| Accumulated interests |
0 | 1,134 | ||||||||||
| Share based incentive |
2,605 | 174 | ||||||||||
|
|
|
|
|
|
|
|||||||
| Total adjustments |
2,962 | 1,897 | ||||||||||
|
|
|
|
|
|
|
|||||||
11. STATEMENT OF CASH FLOWS — CHANGE IN WORKING CAPITAL
| Net change in receivables |
555 | 1,357 | ||||||
| Net change in inventory |
1,126 | 125 | ||||||
| Net change in current debt |
(5,287 | ) | (1,118 | ) | ||||
|
|
|
|
|
|||||
| Total change in working capital |
(3,289 | ) | 364 | |||||
|
|
|
|
|
12. Significant events occurring after the balance sheet date
As of 15 July 2016 Serendex Pharmaceuticals A/S has signed a business agreement with Savara Inc, Texas (USA), including transfer of the entire operation and all activities.
Serendex Pharmaceuticals A/S has
| • | changed its chairman of the board as of 16 July 2016. |
| • | changed its CEO as of 16 July 2016. |
| • | changed its name to Serenova A/S as of 5 September 2016. |
| • | changed its business address as of 2 September 2016. |
No other significant events have occurred subsequent to the balance sheet date that are considered to have a material influence in the evaluation of the 30 June 2016 Interim Report.
13. CONTINGIENT LIABILITIES
Pending litigations
As of 30 June 2016 Serendex has made an external legal assessment of alleged claims against the company. None has been seen as having any material impact — hence, no provisions have been made.
Joint Taxation
Serendex A/S is part of a Danish joint taxation scheme with Sorana A/S, Drugrecure ApS and Pharmaorigin, and has consequently a joint and several liabilities with respect to corporate income taxes etc. for the jointly-taxed companies, and a joint and several liability with respect to any obligations to withhold tax on interest, royalties and dividends for these companies.
14. ACCOUNTING POLICIES
Accounting policies applied in the preparation of the consolidated financial statements are set out below. The accounting policies are unchanged compared to 2015.
163
Table of Contents
New standards and interpretations
Based on an assessment of new or amended and revised accounting standards and interpretations (‘IFRSs’) issued by the International Accounting Standards Board (IASB) effective on or after 1 January 2016, it has been assessed that the application of these new IFRSs has not had a material impact on the Consolidated financial statements in 2016, and Management does not anticipate any significant impact on future periods from the adoption of these new IFRSs.
IASB has issued a number of new or amended and revised accounting standards and interpretations that have not yet come into effect. In general, the following standards are expected to have the most significant impact on current accounting regulation:
| • | IASB has issued IFRS 9 ‘Financial Instruments’, with effective date 1 January 2018. The Management does currently not see this amendment relevant for Serendex Pharmaceuticals A/S. |
| • | IASB has issued IFRS 15 ‘Revenue from contracts with customers’, with effective date 1 January 2018. The Management does currently not see this amendment relevant for Serendex Pharmaceuticals A/S. |
| • | IASB has issued IFRS 16 ‘Leases’ with effective date 1 January 2019. The Management does currently not see this amendment relevant for Serendex Pharmaceuticals A/S. |
Basis of preparation
The Annual Report has been prepared in accordance with the International Financial Reporting Standards (IFRS) as issued by IASB.
The preparation of financial statements in conformity with IFRS requires the use of certain critical accounting estimates. It also requires management to exercise its judgement in the process of applying the Serendex Pharmaceuticals A/S Group’s accounting policies. The areas involving a higher degree of judgment or complexity, and areas where assumptions and estimates are significant to the consolidated financial statements are disclosed.
Serendex has a satisfactory cash situation for the next 12 months – hence the interim report has been prepared for on-going business.
The consolidated financial statements are presented in DKK, reflecting the company’s functional currency.
Basis of consolidation
The consolidated financial statements are prepared by adding the financial statements of the parent company and the individual subsidiaries, all of which are prepared in accordance with the group’s accounting policies.
The following companies are consolidated:
| • | Serendex Pharmaceuticals A/S (parent) |
| • | Drugrecure ApS (100% Serendex) |
| • | Pharmaorigin ApS (100% Serendex) |
Recognition and measurement in general
The net revenue is recognised in the profit and loss account if delivery and risk transfer to the buyer have taken place before the end of the year, and if the income can be determined reliably and is expected to be received. The net revenue is recognised exclusive of VAT and taxes and with the deduction of any discounts granted in
164
Table of Contents
connection with the sale. Recognition of value adjustments of assets and liabilities are recognised in the profit and loss account upon financial assessment.
All costs — including depreciation, amortisation, write-down, provisions, and reversals, which are due to changes in estimated amounts previously recognised in the profit and loss account — are recognised in the profit and loss account.
Assets are recognised in the balance sheet when the company is liable to achieve future, financial benefits and the value of the asset can be measured reliably.
Liabilities are recognised in the balance sheet when the company is liable to lose future, financial benefits and the value of the liability can be measured reliably.
Translation of foreign currency
Operational transactions in foreign currency are translated by using the exchange rate at cost basis upon bank transaction. Differences in the rate of exchange arising between the rate at the date of transaction and the rate at the date of payment are recognised in the profit and loss account as an item under net financials.
Debtors, creditors, and other monetary items in foreign currency — not settled at the date of the balance sheet — are translated by using the period closing rate held by The Danish Central Bank. The difference between the closing rate and the rate at the time of establishment of the receivable or the payable is recognised in the profit and loss account under financial income and financial costs.
Fixed assets and other non-monetary assets acquired in foreign currency and which are not considered to be investment assets purchased in foreign currencies are measured at the exchange rate on the transaction date.
INCOME STATEMENT
Net revenue
As of 30 June 2016 Serendex has only one segment according to IFRS.
Revenue represents amounts receivable for products or services delivered in the normal course of business of the company. Revenue is reduced for estimated customer returns and other similar allowances whenever applicable based on historical data and expectations of future sales. Revenue is recognised upon invoiced sale and when risks and rewards of ownership is transferred to the customer. The risks and rewards of ownership are generally transferred at the time the product is shipped and delivered to the customer. Revenue is recognised in the profit and loss account when management has established that all aforementioned conditions for revenue recognition have been met.
Other operating income and costs comprise accounting items of secondary nature in proportion to the principal activities of the enterprise.
Up-front payments that are attributable to subsequent research and development activities are recognised as deferred revenue and will subsequently be recognised as revenue over the expected contract period. Non-refundable up-front payments and milestone payments that are not attributable to subsequent research and/or development activities or other delivery obligations are recognised as revenue when the contracts are signed or when the milestone criteria are met respectively.
Cost of goods sold
The cost of goods sold comprises costs paid for manufacturing in order to generate net revenue for the year including depreciation, amortisation and write-downs of inventory.
165
Table of Contents
Staff expenses
Staff expenses comprise total remuneration to Serendex employees including fees to Board of Directors.
Raw materials and consumables used
Raw materials and consumables used comprise handling charges, distributions costs and costs paid for manufacturing samples and references.
External expenses
External expenses compromise all external costs including development costs, which are not directly attributable to the Company’s development of new products (capitalised costs). External expenses includes depreciation and write-downs.
The classification of costs (income statement vs. equity), associated with the rights issue is in accordance with IAS32. I.e. costs directly attributable to issuing shares or expected Addition of equity are deducted from equity and costs related to the stock market listing, or otherwise not incremental and directly attributable to issuing new shares, are recognised as an external expense in the income statement.
NET FINANCIALS
Net financials include interest income, interest expenses on loans, and realized and unrealized exchange rate gains and losses. Net financials are recognised in the profit and loss account with the amounts concerning the financial year.
Tax
Tax comprises the current tax for the year and the changes in deferred tax. Tax costs are recognised in the profit and loss account with the amounts concerning the fiscal year with the share referring to entries in the equity subsequently deferred tax asset.
BALANCE SHEET ITEMS
Intangible assets
Intangible fixed assets comprise development projects, patents, and licenses. Development costs comprise costs directly and indirectly attributable to development of new products from which the Company expects a future economic benefit. All other development costs are recognised as costs in the profit and loss accounts.
Capitalised development costs are measured at cost with deduction of accrued amortisations or at the recoverable value, if this is lower.
The carrying amounts of intangible assets carried at cost or amortized cost are tested annually to determine whether there are indications of any impairment in excess of that expressed in normal amortisation. If that is the case, the asset is written down to the recoverable amount, which is the higher value of the net sales price and the capitalised value. Impairment losses on intangible assets are recognised under the same line item as amortisation of the assets. For development projects in progress, the recoverable amount is assessed annually, regardless of whether any indications of impairment have been found.
After completion of the development work, capitalised development costs are amortized on a straight-line basis over the estimated financial useful life.
166
Table of Contents
Profit and loss from the realization of development projects, patents, and licenses are measured as the difference between the sales price with deduction of sales costs and the book value at the time of the sale.
Tangible assets
Tangible assets are measured at cost with deduction of accrued depreciation and write-down. The basis of depreciation is costs with deduction of expected residual value after the end of the useful life of the asset.
The cost comprises the Addition cost and costs directly attached to the Addition until the time when the asset is ready for use. Depreciation takes place on a straight line basis and based on an evaluation of the expected useful life:
Office equipment and fittings: 3 years
IT and software licenses: 2 years
Leasehold improvements: 10 years
Minor assets with an expected useful life of less than 1 year and/or of a cost less than EUR 2,000 (app. DKK 15,000) are recognised as costs in the profit and loss account in the year of Addition.
Profit or loss deriving from the sales of tangible fixed assets is measured as the difference between the sales price reduced by the selling costs and the book value at the time of the sale. Profit or loss is recognised in the profit and loss account under depreciation.
Write-down of assets
The book values of intangible as well as tangible fixed assets are subject to annual write-down assessment in order to disclose any indications of impairment beyond those expressed by amortisation and depreciation respectively.
If indications of impairment are disclosed, impairment tests are carried out for each individual asset or group of assets respectively. Write-down takes place to the recoverable amount, if this value is lower than the book value.
The recoverable value is equal to the value of the net selling price or the value in use, whichever is higher. The value in use is determined as the present value of the expected net income deriving from the use of the asset or the group of assets. Any loss based on the write-down test is recognised in the profit and loss account under depreciation.
Inventories
Inventories are measured at cost on basis of measured average prices. In case the net realizable value is lower than the cost, write-down takes place at this lower value.
The inventory includes:
Addition of pharmaceutical ingredients, which include the cost for raw materials and the initial processing
The cost for manufactured goods and works in progress
The net realizable value for inventories is recognised as the market price with the deduction of completion costs and selling costs, and it is determined by taking negotiability, obsolescence, and the development of the expected market price into consideration. All logistic costs related to the inventories are recognised in the profit and loss account.
167
Table of Contents
Deferred tax
Long-term deferred tax (+12 months) and current tax (less than 12 months) are recognised in the balance sheet at the amount calculated on the basis of the expected taxable income for the year adjusted for tax on previous years, taxable income and prepaid taxes. Tax receivable and tax liabilities are set off to the extent that legal right of set-off exists and if the items are expected to be settled net or simultaneously. Deferred tax is measured on the basis of all temporary differences in assets and liabilities with a balance sheet focus.
Deferred tax is measured based on the tax rules and tax rates applying under the legislation on the balance sheet date and prevailing when the deferred tax is expected to be released as current tax. In the period 2014 to 2016, the corporate tax rate will be reduced gradually from 25% to 22%, which will affect the deferred tax liabilities and deferred tax assets. Unless a recognition with a different tax rate than 22% will result in a significant material deviation in the estimated deferred tax liability or tax asset, deferred tax liabilities and assets are recognised by 22%.
Trade receivables
Trade receivables are recognised at amortized cost less potential losses on doubtful debts. Write-downs are based on individual assessments of each debtor.
Other receivables, prepayments and accrued expenses
Deposits comprise rental deposits paid to real estate agencies.
Prepaid expenses paid in advance but which has not yet been incurred are recognised under assets.
Accrued expenses recognised under assets comprise incurred costs concerning the next financial year.
Cash and cash equivalents
Cash and cash equivalents includes cash in Danske Bank.
Non-current liabilities
Non-current liabilities comprise long term loans to management and corresponds to the outstanding debt of the loan.
Current liabilities
Current liabilities are measured at amortized costs, which usually corresponds to the nominal value.
CASH FLOW STATEMENT
The cash flow statement shows the cash flow of the company for the year, divided in cash flows deriving from Operating activities, Capitalised activities, Financing activities, Changes in the liabilities, Available funds at the beginning and the end of the year respectively
Cash flow from operating activities
Cash flow from operating activities is calculated as the profit and loss results for the year adjusted for non-cash operating items, the change in the working capital, and corporate tax paid/received.
168
Table of Contents
Cash flow from capitalised activities
Cash flow from investment activities comprises development costs directly attributable to the Company’s research and development of new products and payments in connection with the Addition tangible assets.
Cash flow from financing activities
Cash flow from financing activities comprises changes in the size or the composition of the share capital and the costs in this connection. Furthermore, these activities comprise borrowings, instalments on interest bearing debt, and payment of dividend to the shareholders.
15. STATEMENT OF THE BOARD OF DIRECTORS
The Board of Directors on the February 7, 2017 approved the unaudited consolidated financial statements of Serendex A/S for the period 1 January 2016 to 30 June 2016.
169
Table of Contents
(b) Pro Forma Financial Information
UNAUDITED PRO FORMA CONDENSED COMBINED FINANCIAL STATEMENTS
The following unaudited pro forma condensed combined financial statements give effect to the merger between Mast and Savara and Savara’s previously consummated acquisition of Serendex A/S (“Serendex”) as discussed below. The merger is structured as a reverse merger and Savara was determined to be the accounting acquirer based upon the terms of the merger and other factors including: (i) Savara security holders will own approximately 77% of the combined company immediately following the closing of the merger, (ii) Savara directors will hold the majority (5 out of 7) of board seats in the combined company, and (iii) Savara management will hold all key positions in the management of the combined company. The transaction will be accounted for under the acquisition method of accounting under accounting principles generally accepted in the United States (US GAAP). Under the acquisition method of accounting for the purpose of these unaudited pro forma condensed combined financial statements, management of Mast and Savara have determined a preliminary estimated purchase price, calculated as described in Note 2 to these unaudited pro forma condensed combined financial statements. The net tangible and intangible assets acquired and liabilities assumed in connection with the transaction are recorded at their estimated acquisition date fair values. Any excess of purchase price over fair value of identified assets acquired and liabilities assumed will be recognized as goodwill. A final determination of these estimated fair values will be based on the actual net tangible and intangible assets of Mast that exist as of the date of completion of the transaction.
Previously Consummated Serendex Acquisition
On July 15, 2016, Savara completed its acquisition of Serendex for total purchase consideration of $12.4 million. The purchase consideration consisted primarily of $2.9 million in common stock and $9.5 of contingent consideration. The acquisition of Serendex is reflected in Savara’s historical consolidated balance sheet at December 31, 2016.
Pro Forma Information
The unaudited pro forma condensed combined balance sheet as of December 31, 2016 and the unaudited pro forma condensed combined statements of operations for the year ended December 31, 2016 are based on (i) the historical consolidated results of operations of Savara and its subsidiaries (which include the results of Serendex subsequent to Savara’s July 15, 2016 acquisition of Serendex); (ii) the historical consolidated results of operations of Mast; (iii) and the historical results of operations of Serendex for the period January 1, 2016 to July 14, 2016.
The unaudited pro forma condensed combined balance sheet as of December 31, 2016 assumes that the merger took place on December 31, 2016 and combines the historical balance sheets of Mast and Savara as of December 31, 2016. The unaudited pro forma condensed combined statements of operations for the year ended December 31, 2016 assumes that both the merger and the acquisition of Serendex took place as of January 1, 2016, and combines the historical results of Mast and Savara and the pre-acquisition historical results of Serendex. The historical financial statements of Mast, Savara and Serendex (for the interim period through June 30, 2016), which are provided elsewhere in this proxy statement/prospectus/information statement, have been adjusted to give pro forma effect to events that are (i) directly attributable to the mergers, (ii) factually supportable, and (iii) with respect to the statements of operations, expected to have a continuing impact on the combined results.
The unaudited pro forma condensed combined financial statements are based on the assumptions and adjustments that are described in the accompanying notes. The unaudited pro forma condensed combined financial statements and pro forma adjustments have been prepared based on preliminary estimates of fair value of assets acquired and liabilities assumed. Differences between these preliminary estimates and the final acquisition accounting will occur and these differences could have a material impact on the accompanying unaudited pro forma condensed combined financial statements and the combined company’s future results of
170
Table of Contents
operations and financial position. The actual amounts recorded as of the completion of the merger may differ materially from the information presented in these unaudited pro forma condensed combined financial statements as a result of the amount, if any, of capital raised by Savara between entering the Merger Agreement and closing of the merger; the amount of cash used by Mast’s operations between the signing of the Merger Agreement and the closing of the merger; the timing of closing of the merger; and other changes in the Mast assets and liabilities that occur prior to the completion of the merger.
The unaudited pro forma condensed combined financial statements do not give effect to the potential impact of current financial conditions, regulatory matters, operating efficiencies or other savings or expenses that may be associated with the acquisition. The unaudited pro forma condensed combined financial statements have been prepared for illustrative purposes only and are not necessarily indicative of the financial position or results of operations in future periods or the results that actually would have been realized had Mast, Savara and Serendex been a combined company during the specified periods. The unaudited pro forma condensed combined financial statements, including the notes thereto, should be read in conjunction with the audited financial statements of Mast and Savara for the year ended December 31, 2016 and the unaudited condensed financial statements of Serendex for the six months ended June 30, 2016 included elsewhere in this proxy statement/prospectus/information statement.
171
Table of Contents
Unaudited Pro Forma Condensed Combined Balance Sheet
December 31, 2016
(in thousands)
| Mast | Savara | Pro Forma Merger Adjustments |
Pro Forma Combined |
|||||||||||||||||
| Assets |
||||||||||||||||||||
| Current assets: |
||||||||||||||||||||
| Cash and cash equivalents |
$ | 8,542 | $ | 13,373 | $ | — | $ | 21,915 | ||||||||||||
| Investment securities |
2,740 | — | — | 2,740 | ||||||||||||||||
| Grant and awards receivable |
— | 400 | 400 | |||||||||||||||||
| Prepaid expenses and other assets |
903 | 840 | — | 1,743 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total current assets |
12,185 | 14,613 | — | 26,798 | ||||||||||||||||
| Property, plant, and equipment, net |
99 | 793 | — | 892 | ||||||||||||||||
| In-process research and development |
2,500 | 10,477 | 18,803 | G | 31,780 | |||||||||||||||
| Goodwill |
3,007 | 3,051 | 9,517 | H | 15,575 | |||||||||||||||
| Deposits and other non-current assets |
131 | — | — | 131 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total assets |
$ | 17,922 | $ | 28,934 | $ | 28,320 | $ | 75,176 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Liabilities, redeemable convertible preferred stock and stockholders’ deficit |
|
|||||||||||||||||||
| Current liabilities: |
||||||||||||||||||||
| Accounts payable |
$ | 626 | $ | 536 | $ | — | $ | 1,162 | ||||||||||||
| Accrued expenses and other liabilities |
1,974 | 2,477 | 3,482 | D | 9,783 | |||||||||||||||
| 1,850 | E | |||||||||||||||||||
| Accrued compensation and payroll taxes |
718 | — | — | 718 | ||||||||||||||||
| Debt facility |
1,548 | — | — | 1,548 | ||||||||||||||||
| Capital lease obligation, current portion |
— | 442 | — | 442 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total current liabilities |
4,866 | 3,455 | 5,332 | 13,653 | ||||||||||||||||
| Noncurrent liabilities: |
||||||||||||||||||||
| Accrued interest on convertible promissory notes |
— | 151 | (151 | ) | F | — | ||||||||||||||
| Debt facility, net of current portion |
2,285 | — | — | 2,285 | ||||||||||||||||
| Deferred income tax liability |
995 | 2,305 | 7,526 | I | 10,826 | |||||||||||||||
| Convertible promissory notes |
— | 3,448 | (3,448 | ) | F | — | ||||||||||||||
| Put option liability |
— | 979 | (979 | ) | F | — | ||||||||||||||
| Contingent consideration |
— | 9,708 | — | 9,708 | ||||||||||||||||
| Capital lease obligation, net of current portion |
17 | 579 | — | 596 | ||||||||||||||||
| Other long-term liabilities |
— | 323 | — | 323 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total liabilities |
8,163 | 20,948 | 8,280 | 37,391 | ||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Redeemable convertible preferred stock: |
||||||||||||||||||||
| Convertible preferred stock |
— | 43,861 | (43,861 | ) | C | — | ||||||||||||||
| Stockholders’ equity: |
||||||||||||||||||||
| Common stock |
255 | 5 | 222 | B | 1,017 | |||||||||||||||
| 490 | C | |||||||||||||||||||
| 45 | F | |||||||||||||||||||
| Additional paid-in-capital |
320,576 | 3,117 | (290,278 | ) | A | 82,184 | ||||||||||||||
| (222 | ) | B | ||||||||||||||||||
| 43,371 | C | |||||||||||||||||||
| 5,620 | F | |||||||||||||||||||
| Accumulated other comprehensive income/(loss) |
1 | (591 | ) | (1 | ) | A | (591 | ) | ||||||||||||
| Accumulated earnings/(deficit) |
(311,073 | ) | (38,406 | ) | 311,073 | A | (44,825 | ) | ||||||||||||
| (3,482 | ) | D | ||||||||||||||||||
| (1,850 | ) | E | ||||||||||||||||||
| (1,087 | ) | F | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total stockholders’ equity/(deficit) |
9,759 | (35,875 | ) | 63,901 | 37,785 | |||||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total liabilities and stockholders’ equity |
$ | 17,922 | $ | 28,934 | $ | 28,320 | $ | 75,176 | ||||||||||||
|
|
|
|
|
|
|
|
|
|||||||||||||
See accompanying notes to the unaudited pro forma condensed combined financial statements.
172
Table of Contents
Unaudited Pro Forma Condensed Combined Statement of Operations
(in thousands, except share and per share data)
| For the Year Ended December 31, 2016 |
||||||||||||||||||||||||
| Mast | Savara | Serendex | Pro Forma Merger Adjustment |
Pro Forma Combined |
||||||||||||||||||||
| (see note 4) | ||||||||||||||||||||||||
| Grant revenue |
$ | 128 | $ | 400 | $ | — | $ | — | $ | 528 | ||||||||||||||
| Operating expenses: |
||||||||||||||||||||||||
| Product development |
20,793 | 8,182 | 4,102 | — | 33,077 | |||||||||||||||||||
| General and administrative |
9,342 | 2,503 | 2,665 | — | 14,510 | |||||||||||||||||||
| Impairment of IPR&D |
6,049 | — | — | — | 6,049 | |||||||||||||||||||
| Depreciation and amortization |
99 | 346 | — | — | 445 | |||||||||||||||||||
| Transaction related costs |
301 | 317 | — | (618 | ) | J | — | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Total operating expenses |
36,584 | 11,348 | 6,767 | (618 | ) | 54,081 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Loss from operations |
(36,456 | ) | (10,948 | ) | (6,767 | ) | 618 | (53,553 | ) | |||||||||||||||
| Interest and other income (expense), net |
(2,053 | ) | (332 | ) | (58 | ) | — | (2,443 | ) | |||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Loss before income taxes |
(38,509 | ) | (11,280 | ) | (6,825 | ) | 618 | (55,996 | ) | |||||||||||||||
| Income taxes |
2,409 | 357 | — | — | 2,766 | |||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Net loss |
$ | (36,100 | ) | $ | (10,923 | ) | $ | (6,825 | ) | $ | 618 | $ | (53,230 | ) | ||||||||||
| Accretion of preferred stock classified as mezzanine equity |
— | (94 | ) | — | — | (94 | ) | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Net loss attributable to common stockholders |
$ | (36,100 | ) | $ | (11,017 | ) | $ | (6,825 | ) | $ | 618 | $ | (53,324 | ) | ||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Basic and diluted net loss per share |
$ | (0.17 | ) | $ | (3.29 | ) | $ | — | $ | — | $ | (0.06 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
| Weighted average common share outstanding- basic and diluted |
208,484,370 | 3,348,647 | — | 676,491,896 | B | 888,324,913 | ||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||||
See accompanying notes to the unaudited pro forma condensed combined financial statements.
173
Table of Contents
NOTES TO THE UNAUDITED PRO FORMA CONDENSED
COMBINED FINANCIAL INFORMATION
1. Description of Transaction and Basis of Presentation
Description of Transaction
On January 6, 2017, Savara entered into the Merger Agreement with Mast, pursuant to which, among other things, subject to the satisfaction or waiver of the conditions set forth in the Merger Agreement, that a wholly-owned subsidiary of Mast will merge with and into Savara, with Savara becoming a wholly-owned subsidiary of Mast and the surviving corporation of the merger. At the closing of the merger, each outstanding share of Savara’s common stock will be converted into the right to receive approximately 41 pre-split shares of common stock of Mast (before giving effect to the Reverse Stock Split), as well as the payment of cash in lieu of fractional shares. Immediately following the effective time of the merger, Mast equity holders are expected to own approximately 23% of the outstanding capital stock of the combined company, with Savara’s preexisting equity holders expected to own approximately 77%. Note that share references in these pro forma condensed combined financial statements do not include the effects of the proposed Reverse Stock Split (discussed in the section entitled “Mast Proposal No. 2”).
Basis of Presentation
The unaudited pro forma condensed combined financial statements were prepared in accordance with the regulations of the Securities and Exchange Commission (SEC). The unaudited pro forma condensed combined balance sheet as of December 31, 2016 is presented as if the merger had been completed on December 31, 2016. The unaudited pro forma condensed combined statement of operations for the year ended December 31, 2016 assumes that both the merger and Savara’s acquisition of Serendex took place as of January 1, 2016, and combines the historical results of Mast and Savara and the pre-acquisition historical results of Serendex.
Based on the terms of the merger, Savara is deemed to be the acquiring company for accounting purposes and the merger will be accounted for under the acquisition method of accounting in accordance with the provisions of Accounting Standards Codification 805, Business Combinations. Accordingly, assets and liabilities of Savara will be recorded as of the merger closing date at their respective carrying value and assets and liabilities of Mast will be recorded as of the merger closing date at their respective fair values. Under the acquisition method of accounting for the purpose of these unaudited pro forma financial statements, management of Savara and Mast have determined a preliminary estimated purchase price, calculated as described in Note 2 to these unaudited pro forma condensed combined financial statements. The net tangible assets acquired and liabilities assumed in connection with the transaction are at their estimated acquisition date fair values. A final determination of these estimated fair values will be based on the actual net tangible assets of Mast that exist as of the date of completion of the transaction.
To the extent there are significant changes to the business following completion of the merger, the assumptions and estimates set forth in the unaudited pro forma condensed combined financial statements could change significantly. Accordingly, the pro forma purchase price adjustments are subject to further adjustments as additional information becomes available and as additional analyses are conducted following the completion of the merger. There can be no assurances that these additional analyses will not result in material changes to the estimates of fair value.
2. Preliminary Purchase Price
The preliminary estimated purchase price of the merger is $30.6 million using Mast’s share price for its common stock and its common shares outstanding as of the close of business on March 6, 2017. Note that in a reverse merger, the purchase consideration determined under US GAAP will be based on the market capitalization of Mast on the date of the merger. The estimated fair value of the net assets acquired, excluding goodwill is $18.0 million.
174
Table of Contents
Management of Savara has preliminarily concluded the proposed merger is a business combination and will apply the acquisition method of accounting. Under the acquisition method of accounting, the total purchase price is allocated to the acquired tangible and intangible assets and assumed liabilities of Mast based on their estimated fair values as of the proposed merger closing date. The excess of the purchase price over the fair value of assets acquired and liabilities assumed, if any, is allocated to goodwill. To the extent the actual purchase price varies from the estimated purchase price used in these unaudited pro forma condensed combined financial information, the impact will be an increase or decrease in goodwill.
The preliminary allocation of the estimated total purchase price of the proposed merger is as follows (in thousands):
| Fair value of Mast net assets to carry over to merged company |
$ | 18,046 | ||
| Goodwill |
12,524 | |||
|
|
|
|||
| Total purchase consideration |
$ | 30,570 | ||
|
|
|
The preliminary estimated fair values of the acquired assets and assumed liabilities of Mast as of December 31, 2016 is as follows (in thousands):
| Net tangible assets |
$ | 5,264 | ||
| In-process research and development intangible asset, net of deferred tax liability |
12,782 | |||
|
|
|
|||
| Estimated fair value of net assets acquired |
$ | 18,046 | ||
|
|
|
Note that while the purchase accounting assuming the merger occurred on December 31, 2016 reflects positive net tangible assets, Mast continues to fund its operations through the close of the merger with cash on hand. As such, Savara does not expect to acquire any substantive amount of cash upon consummation of the merger. The allocation of the estimated purchase price is preliminary because the proposed merger has not yet been completed. The purchase price allocation will remain preliminary until Savara’s management determines the fair values of assets acquired and liabilities assumed. The final determination of the purchase price allocation is anticipated to be completed as soon as practicable after completion of the merger and will be based on the fair values of the assets acquired and liabilities assumed as of the merger closing date. The final amounts allocated to assets acquired and liabilities assumed could differ significantly from the amounts presented in the unaudited pro forma condensed combined financial statements.
3. Pro Forma Adjustments
Pro forma adjustments are necessary to reflect the acquisition consideration exchanged and to adjust amounts related to the tangible assets and liabilities of Mast to reflect the preliminary estimate of their fair values, and to reflect the impact on the statements of operations of the merger as if the companies had been combined during the periods presented therein. The pro forma adjustments included in the unaudited pro forma condensed combined financial statements are as follows:
| A. | To reflect the elimination of Mast’s historical stockholders’ equity balances, including accumulated deficit and accumulated other comprehensive income, and to reflect the adjustments to the fair value of Mast’s net assets recorded in the preliminary allocation of the estimated total purchase price, at the close of the merger referred to in Note 2 above. |
| Elimination of Mast’s accumulated deficit |
$ | (311,073 | ) | |
| Elimination of Mast’s accumulated other comprehensive income |
1 | |||
| Fair value adjustment to intangible assets (see G below) |
18,803 | |||
| Fair value adjustment to goodwill (see H below) |
9,517 | |||
| Adjustment to deferred tax liability (see I below) |
(7,526 | ) | ||
|
|
|
|||
| Total |
$ | (290,278 | ) | |
|
|
|
175
Table of Contents
| B. | To reflect the reclassification Savara’s par value of common stock and additional paid-in capital in connection with the exchange of Savara’s common stock for Mast’s common stock. |
| C. | To reflect the conversion of Savara’s redeemable convertible preferred stock to Mast common stock. |
| D. | To record $3.5 million of estimated transaction costs that were not incurred as of December 31, 2016. |
| E. | To record $1.9 million of severance liabilities in relation to termination of employees of Mast upon consummation of the merger. |
| F. | To reflect the conversion of $4.4 million in aggregate principal of, and accrued interest on, Savara’s convertible notes into approximately 1.1 million shares of Savara common stock and then into shares of Mast common stock and to reflect the elimination of the put option (redemption feature) on Savara’s convertible notes. |
| G. | To record intangible assets acquired in the merger and eliminate Mast’s historical intangible assets. |
| To record intangible assets acquired in the merger |
$ | 21,303 | ||||||
| To eliminate historical Mast intangible assets |
(2,500 | ) | ||||||
|
|
|
|||||||
| Total |
$ | 18,803 | ||||||
|
|
|
| H. | To record goodwill as a result of the merger and eliminate Mast’s historical goodwill. |
| To record goodwill acquired in the merger |
$ | 12,524 | ||||||
| To eliminate historical Mast goodwill |
(3,007 | ) | ||||||
|
|
|
|||||||
| Total |
$ | 9,517 | ||||||
|
|
|
| I. | To eliminate Mast’s deferred tax liability related to prior acquisitions that arose from amortizing, for tax purposes, intangible assets from business combination transactions prior to this merger and record deferred tax liability related to the merger (assumes a 40% tax rate applied to intangible assets acquired). |
| To record net deferred tax liability related to the merger |
$ | 8,521 | ||||||
| To eliminate deferred tax liabilities related to Mast’s intangible assets from prior acquisitions |
(995 | ) | ||||||
|
|
|
|||||||
| Total |
$ | 7,526 | ||||||
|
|
|
| J. | To eliminate nonrecurring transaction costs incurred during the year ended December 31, 2016 of $0.6 million that are directly related to the merger. |
176
Table of Contents
4. Serendex’s Historical Financial Statements
Schedule 1
Serendex
Statements of Operations
For the Period from January 1, 2016 to July 14, 2016
| January 1, 2016 to June 30, 2016 (1)(2) |
July 1, 2016 to July 14, 2016 (1) |
US GAAP Adjustments |
As Converted to US GAAP |
|||||||||||||||||||
| DKK | DKK | DKK | DKK | USD | ||||||||||||||||||
| Grant revenue |
704 | — | (704 | ) | (a) | — | $ | — | ||||||||||||||
| Operating expenses |
||||||||||||||||||||||
| Product development |
6,034 | 3,336 | 18,014 | (a) (b) | 27,384 | 4,102 | ||||||||||||||||
| General and administrative |
12,954 | 4,834 | — | 17,788 | 2,665 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Total operating expenses |
18,988 | 8,170 | 18,014 | 45,172 | 6,787 | |||||||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Loss from operations |
(18,284 | ) | (8,170 | ) | (18,718 | ) | (45,172 | ) | (6,787 | ) | ||||||||||||
| Interest and other income (expense), net |
(328 | ) | (59 | ) | — | (387 | ) | (58 | ) | |||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| Net loss |
(18,612 | ) | (8,229 | ) | (18,718 | ) | (45,559 | ) | $ | (6,825 | ) | |||||||||||
|
|
|
|
|
|
|
|
|
|
|
|||||||||||||
| (1) | Amounts derived from Serendex’s accounting records in accordance with the IFRS issued by the IASB and have been reclassified to be consistent with the manner in which items are classified in Savara consolidated statement of operations and comprehensive loss. |
| (2) | Amounts derived from Serendex’s historical unaudited condensed financial statements for the six months ended June 30, 2016 included or incorporated by reference elsewhere in this proxy statement/prospectus/information statement. |
US GAAP Adjustments to Serendex’s Historical Financial Statements
On July 15, 2016, Savara completed the acquisition of Serendex through its wholly-owned subsidiary, Savara ApS. Serendex prepared its financial statements in accordance with the IFRS issued by the IASB. Included in Schedules 1 above are the US GAAP adjustments to Serendex’s historical financial statements for the period from January 1, 2016 to July 14, 2016.
(a) Revenue recognition
| i. | In accordance with the IFRS issued by the IASB, revenue generated from sales of active pharmaceutical ingredient (API) to vendors associated with clinical trial studies is recognized as net revenue on the financial statements. |
| ii. | Under US GAAP, revenue generated from sales of API to vendors associated with clinical trial studies would be considered contra- R&D expenses as those revenues were not generated due to commercialized sales to customers. |
(b) Research and development costs- capitalization
| i. | Under DK GAAP, research and development costs directly and indirectly attributable to development of new products are capitalized as in-process R&D. |
| ii. | Under US GAAP, research and development costs are expensed as incurred. |
177
Table of Contents
Translation of Serendex’s Historical Financial Statements to US Dollars
The unaudited pro forma condensed combined financial information is presented in US dollars unless otherwise stated, and accordingly, the financial information of Serendex used to prepare the unaudited pro forma condensed combined financial information was translated from Danish Krone to US dollars (Schedules 1) using the following exchange rate, which correspond with the exchange rate for the periods being presented:
| Statement of operations for the period from January 1, 2016 to July 14, 2016 (pre-acquisition period): Average for period |
1 = US$ | .1498 |
178
Table of Contents
| (d) | Exhibits. |
| Exhibit No. |
Description | |
| 2.1 | Business Transfer Agreement, dated May 13, 2016, between Savara Inc. and Serendex Pharmaceuticals A/S (Incorporated by reference to Exhibit 2.6 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017) | |
| 3.1 | Amended and Restated Certificate of Incorporation of the Company | |
| 4.1 | Form of Stock Purchase Warrant first issued by Savara Inc. on May 30, 2012 (Incorporated by reference to Exhibit 4.10 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 4.2 | Form of Stock Purchase Warrant first issued by Savara Inc. on July 15, 2016 (Incorporated by reference to Exhibit 4.11 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.1# | Savara Inc. Stock Option Plan (Incorporated by reference to Exhibit 10.53 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.2# | Savara Inc. Form of Incentive Stock Option Agreement (Incorporated by reference to Exhibit 10.54 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.3# | Form of Stock Issuance Agreement (Incorporated by reference to Exhibit 10.55 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.4# | Executive Employment Agreement, dated March 9, 2017, between Savara Inc. and Robert Neville (Incorporated by reference to Exhibit 10.56 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.5# | Executive Employment Agreement, dated March 9, 2017, between Savara Inc. and Taneli Jouhikainen (Incorporated by reference to Exhibit 10.57 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.6# | Executive Employment Agreement, dated March 9, 2017, between Savara Inc. and David Lowrance (Incorporated by reference to Exhibit 10.58 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.7† | Supply Agreement, dated September 26, 2016, between Savara Inc. and Xellia Pharmaceuticals ApS (Incorporated by reference to Exhibit 10.59 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.8† | Supply Agreement, effective September 1, 2012, between Savara Inc. and Plastiape SpA, as amended by Amendment No. 1, dated June 1, 2016 (Incorporated by reference to Exhibit 10.60 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.9† | Supply and Licensing Agreement, dated December 10th, 2012, and Addendum to Supply and License Agreement, dated February 22, 2016, between Savara Inc. and GEMA Biotech S.A. (Incorporated by reference to Exhibit 10.61 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.10† | Commercial Supply Agreement dated April 24, 2015 between PARI Pharma GmbH and Serendex Pharmaceuticals A/S (Incorporated by reference to Exhibit 10.62 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.11† | Research Collaboration and License Agreement dated November 7, 2014 between PARI Pharma GmbH and Serendex Pharmaceuticals A/S (Incorporated by reference to Exhibit 10.63 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.12# | Form of Director and Officer Indemnification Agreement (Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on October 23, 2006, File No. 061156993) | |
| 21.1 | List of Subsidiaries | |
| 23.1 | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm to Savara Inc. | |
| 23.2 | Consent of Grant Thornton LLP, Independent Registered Public Accounting Firm to Savara Inc. | |
| 99.1 | Press Release dated April 27, 2017 | |
179
Table of Contents
| # | Indicates management contract or compensatory plan |
| † | Indicates that confidential treatment has been granted to certain portions, which portions have been omitted and filed separately with the SEC. |
180
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
| Date: April 27, 2017 | SAVARA INC., a Delaware corporation | |||||
| By: | /s/ Robert Neville | |||||
| Robert Neville | ||||||
| Chief Executive Officer | ||||||
181
Table of Contents
EXHIBIT INDEX
| Exhibit |
Description | |
| 2.1 | Business Transfer Agreement, dated May 13, 2016, between Savara Inc. and Serendex Pharmaceuticals A/S (Incorporated by reference to Exhibit 2.6 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017) | |
| 3.1 | Amended and Restated Certificate of Incorporation of the Company | |
| 4.1 | Form of Stock Purchase Warrant first issued by Savara Inc. on May 30, 2012 (Incorporated by reference to Exhibit 4.10 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 4.2 | Form of Stock Purchase Warrant first issued by Savara Inc. on July 15, 2016 (Incorporated by reference to Exhibit 4.11 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.1# | Savara Inc. Stock Option Plan (Incorporated by reference to Exhibit 10.53 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.2# | Savara Inc. Form of Incentive Stock Option Agreement (Incorporated by reference to Exhibit 10.54 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.3# | Form of Stock Issuance Agreement (Incorporated by reference to Exhibit 10.55 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.4# | Executive Employment Agreement, dated March 9, 2017, between Savara Inc. and Robert Neville (Incorporated by reference to Exhibit 10.56 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.5# | Executive Employment Agreement, dated March 9, 2017, between Savara Inc. and Taneli Jouhikainen (Incorporated by reference to Exhibit 10.57 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.6# | Executive Employment Agreement, dated March 9, 2017, between Savara Inc. and David Lowrance (Incorporated by reference to Exhibit 10.58 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.7† | Supply Agreement, dated September 26, 2016, between Savara Inc. and Xellia Pharmaceuticals ApS (Incorporated by reference to Exhibit 10.59 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.8† | Supply Agreement, effective September 1, 2012, between Savara Inc. and Plastiape SpA, as amended by Amendment No. 1, dated June 1, 2016 (Incorporated by reference to Exhibit 10.60 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.9† | Supply and Licensing Agreement, dated December 10th, 2012, and Addendum to Supply and License Agreement, dated February 22, 2016, between Savara Inc. and GEMA Biotech S.A. (Incorporated by reference to Exhibit 10.61 to Amendment No. 1 to the Company’s Registration Statement on Form S-4 filed on March 13, 2017.) | |
| 10.10† | Commercial Supply Agreement dated April 24, 2015 between PARI Pharma GmbH and Serendex Pharmaceuticals A/S (Incorporated by reference to Exhibit 10.62 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.11† | Research Collaboration and License Agreement dated November 7, 2014 between PARI Pharma GmbH and Serendex Pharmaceuticals A/S (Incorporated by reference to Exhibit 10.63 to the Company’s Registration Statement on Form S-4 filed on February 10, 2017.) | |
| 10.12# | Form of Director and Officer Indemnification Agreement (Incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed on October 23, 2006, File No. 061156993) | |
| 21.1 | List of Subsidiaries | |
| 23.1 | Consent of PricewaterhouseCoopers LLP | |
| 23.2 | Consent of Grant Thornton LLP | |
| 99.1 | Press Release dated April 27, 2017 | |
182
Table of Contents
| # | Indicates management contract or compensatory plan |
| † | Indicates that confidential treatment has been granted to certain portions, which portions have been omitted and filed separately with the SEC. |
183
Exhibit 3.1
MAST THERAPEUTICS, INC.
AMENDED AND RESTATED CERTIFICATE OF INCORPORATION
(Pursuant to Sections 242 and 245 of the
General Corporation Law of the State of Delaware)
Mast Therapeutics, Inc., a corporation organized and existing under and by virtue of the provisions of the General Corporation Law of the State of Delaware (the “General Corporation Law”), does hereby certify as follows.
1. The name of this corporation is Mast Therapeutics, Inc. and that that this corporation was originally incorporated pursuant to the General Corporation Law on December 1, 1995 under the name Victoria Enterprises, Inc.
2. The Board of Directors of this corporation duly adopted resolutions proposing to amend and restate the Amended and Restated Certificate of Incorporation of this corporation, declaring said amendment and restatement to be advisable and in the best interests of this corporation and its stockholders, and authorizing the appropriate officers of this corporation to solicit the consent of the stockholders therefor, which resolution setting forth the proposed amendment and restatement is as follows.
RESOLVED, that the Amended and Restated Certificate of Incorporation of this corporation be amended and restated in its entirety to read as set forth on Exhibit A attached hereto and incorporated herein by this reference.
3. Exhibit A referred to above is attached hereto as Exhibit A and is hereby incorporated herein by this reference. This Amended and Restated Certificate of Incorporation was approved by the holders of the requisite number of shares of this corporation in accordance with Section 228 of the General Corporation Law.
4. This Amended and Restated Certificate of Incorporation, which restates and integrates and further amends the provisions of this corporation’s Amended and Restated Certificate of Incorporation, has been duly adopted in accordance with Sections 242 and 245 of the General Corporation Law.
IN WITNESS WHEREOF, this Amended and Restated Certificate of Incorporation has been executed by a duly authorized officer of this corporation on this 27th day of April, 2017.
| By: | /s/ Brian Culley | |
| Brian Culley, Chief Executive Officer |
AMENDED AND RESTATED CERTIFICATE OF INCORPORATION,
OF
MAST THERAPEUTICS, INC.
ARTICLE I
The name of this corporation is Savara Inc. (the “Corporation”).
ARTICLE II
The address of the Corporation’s registered office in the State of Delaware is Corporation Service Company, 2711 Centerville Road, Suite 400, Wilmington, County of New Castle, Delaware 19808. The name of its registered agent at such address is Corporation Service Company.
ARTICLE III
The purpose of the Corporation is to engage in any lawful act or activity for which corporations may be organized under the Delaware General Corporation Law (the “DGCL”).
ARTICLE IV
(A) Classes of Stock. The Corporation is authorized to issue two classes of stock to be designated, respectively, “Common Stock” and “Preferred Stock.” The total number of shares which the Corporation is authorized to issue is Five Hundred One Million shares (501,000,000), each with a par value of $0.001 per share. Five Hundred Million (500,000,000) shares shall be Common Stock, and One Million (1,000,000) shares shall be Preferred Stock.
Upon the close of trading on the NYSE MKT on April 27, 2017 (the “Effective Time”), each seventy (70) shares of the Common Stock, par value $0.001 per share, of the Corporation issued and outstanding or held in treasury at the Effective Time shall be reclassified as and changed into one (1) share of Common Stock, par value $0.001 per share, of the Corporation, without any action by the holders thereof. In lieu of any fractional shares to which a holder of shares of Common Stock of the Corporation would be otherwise entitled, the Corporation shall pay in cash, without interest, an amount equal to such fractional interest (after taking into account and aggregating all shares of Common Stock then held by such holder) multiplied by the closing price of the Common Stock as last reported on the NYSE MKT on the day of the Effective Time (determined on a post-split basis).
(B) Preferred Stock. Except as otherwise provided in any certificate(s) of designations duly filed with the Secretary of State of the State of Delaware, the Board of Directors of the Corporation (the “Board”) is hereby expressly authorized to provide for the issuance, in one or more series, of all or any of the shares of Preferred Stock and to fix or alter the rights, preferences, privileges and restrictions granted to or imposed upon such series of Preferred Stock, and the number of shares constituting any such series and the designations thereof, or of any of them, such designations, preferences, and relative, participating, optional or other rights and such qualifications, limitations, or restrictions thereof, as shall be stated and expressed in the resolution or resolutions adopted by the Board providing for the issuance of such shares and as may be permitted by the DGCL. The rights, privileges, preferences and restrictions of any such series of Preferred Stock may be subordinated to, pari passu with (including, without limitation, inclusion in provisions with respect to liquidation and acquisition preferences, redemption or approval of matters by vote or written consent), or senior to any of those of any present or future class or series of Preferred Stock or Common Stock. The Board is also expressly authorized to increase or decrease the number of shares of any series prior or subsequent to the issue of that series, but not below the number of shares of such series
then outstanding. In case the number of shares of any series shall be so decreased, the shares constituting such decrease shall resume the status which they had prior to the adoption of the resolution originally fixing the number of shares of such series.
ARTICLE V
In furtherance and not in limitation of the powers conferred by statutes, the Board is expressly authorized to make, alter, amend or repeal the Bylaws of the Corporation.
ARTICLE VI
The business and affairs of the Corporation shall be managed by or under the direction of the Board. In addition to the powers and authority expressly conferred upon them by statute or by this Certificate of Incorporation or the Bylaws of the Corporation, the Board is hereby empowered to exercise all such powers and do all such acts and things as may be exercised or done by the Corporation. Elections of members of the Board need not be by written ballot unless otherwise provided in the Bylaws of the Corporation.
ARTICLE VII
(A) To the fullest extent permitted by the DGCL, as the same exists or as may hereafter be amended, a director shall not be personally liable to the Corporation or its stockholders for monetary damages for breach of fiduciary duty as a director.
(B) The Corporation shall indemnify to the fullest extent permitted by law any person made or threatened to be made a party to an action or proceeding, whether criminal, civil, administrative or investigative, by reason of the fact that such person, such person’s testator or intestate is or was a director or officer of the Corporation or any predecessor of the Corporation, or serves or served at any other enterprise as a director or officer of the Corporation at the request of the Corporation or any predecessor to the Corporation.
(C) Neither any amendment nor repeal of this Article VII, nor the adoption of any provision of the Corporation’s Certificate of Incorporation inconsistent with this Article VII, shall eliminate or reduce the effect of this Article VII in respect of any matter occurring, or any action or proceeding accruing or arising or that, but for this Article VII, would accrue or arise, prior to such amendment, repeal or adoption of an inconsistent provision.
ARTICLE VIII
The Corporation reserves the right at any time, and from time to time, to amend, alter, change or repeal any provision contained in this Certificate of Incorporation, and other provisions authorized by the laws of the State of Delaware at the time in force may be added or inserted, in the manner now or hereafter prescribed by law; and all rights, preferences and privileges of whatsoever nature conferred upon stockholders, directors or any other persons whomsoever by and pursuant to this Certificate of Incorporation in its present form or as hereafter amended are granted subject to the rights reserved in this Article VIII.
Exhibit 21.1
| Subsidiary | Jurisdiction of Incorporation | |
| Aires Pharmaceuticals, Inc. |
Delaware | |
| SD Pharmaceuticals, Inc. |
Delaware | |
| Aravas Inc. |
Delaware | |
| Savara ApS |
Denmark |
Exhibit 23.1
CONSENT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
We hereby consent to the incorporation by reference in the Registration Statements on Form S-3 (Nos. 333-202960, 333-188870, 333-174203, 333-164177, 333-133824, 333-127857 and 333-117022) and the Registration Statements on Form S-8 (Nos. 333-126551, 333-151903, 333-174940, 333-190376, 333-198046 and 333-206330) of Savara Inc. (formerly known as Mast Therapeutics, Inc.) of our report dated March 10, 2017 relating to the financial statements of Savara Inc. (now known as Aravas Inc.) which appears in this Form 8-K of Savara Inc.
/s/ PricewaterhouseCoopers LLP
Austin, Texas
April 27, 2017
Exhibit 23.2
CONSENT OF INDEPENDENT AUDITORS
We have issued our report dated February 7, 2017 with respect to the consolidated financial statements of Serendex Pharmaceuticals A/S included in this Current Report on Form 8-K of Savara Inc. (formerly Mast Therapeutics, Inc.). We consent to the incorporation by reference of the said report in the Registration Statement of Savara Inc. on Form S-3 (File No. 333-202960).
| /s/ GRANT THORNTON |
| GRANT THORNTON |
| State-Authorized Public Accountants |
| Copenhagen, Denmark |
| April 27, 2017 |
Exhibit 99.1

SAVARA ANNOUNCES CLOSING OF MERGER WITH MAST THERAPEUTICS
Commences Trading on Nasdaq Capital Market on April 28, 2017 Under Ticker Symbol “SVRA”
Conference Call Scheduled for Tuesday May 2nd, 2017 at 4:30 p.m. ET / 3:30 p.m. CT
AUSTIN, TX – April 27, 2017 – Savara Inc. (NASDAQ: SVRA), a clinical-stage specialty pharmaceutical company focused on the development and commercialization of novel therapies for the treatment of serious or life-threatening rare respiratory diseases, today announced the closing of its previously announced merger with Mast Therapeutics, Inc. (NYSE MKT: MSTX), under which the stockholders of Savara have become the majority owners of Mast, and the operations of Mast and Savara have combined. The post-merger company, named Savara Inc., is based in Austin, TX and features three inhaled product candidates, each in advanced stages of clinical development. The company will be led solely by Savara’s current management team. Two independent members of the Mast board remain on the post-merger board together with all five members of the Savara board. Savara’s common stock will commence trading on April 28th, 2017 on the Nasdaq Capital Market under the trading symbol “SVRA”.
“Savara’s transition to the public market marks a significant milestone for us, and serves as testament to the determination of our team as well as the support of our investors to date,” stated Rob Neville, Chairman and CEO of Savara. “Savara’s team is passionate about helping those who suffer from rare and debilitating lung diseases and will dynamically pursue opportunities to develop impactful products to treat such conditions. We believe Savara presents an attractive business opportunity with our pipeline of unique products with considerable market potential, as well as significant value-driving clinical milestones.”
Savara began the development of AeroVanc in 2010 and is now in preparation for a pivotal Phase 3 study. In July 2016, Savara acquired Serendex Pharmaceuticals adding Molgradex to its pipeline. Molgradex is currently in Phase 2/3 development. With the closing of the Mast merger, Savara adds the Aironite program to its pipeline (also known as AIR001). Savara intends to continue its growth strategy focused on indication expansion, strategic development partnerships and product acquisitions.
In connection with the closing of the merger, Mast effected a 1 for 70 reverse split of its common stock. Post-merger and post-reverse split, Savara has approximately 15 million shares of common stock issued and outstanding with prior Savara stockholders collectively owning approximately 77% of the combined company, and prior Mast stockholders collectively owning approximately 23% of the combined company. Prior to the merger closing, Savara stockholders exercised certain previously issued warrants to purchase Savara shares and invested additional capital into the company, resulting in aggregate net proceeds of approximately $4 million.
Savara’s pipeline now includes:
| • | Molgradex, an inhaled nebulized GM-CSF to treat pulmonary alveolar proteinosis (PAP) currently in Phase 2/3 development; |
| • | AeroVanc, an inhaled dry-powder vancomycin to treat chronic methicillin-resistant Staphylococcus aureus (MRSA) pulmonary infection in cystic fibrosis (CF) in preparation for a pivotal Phase 3 study; and |
| • | Aironite, an inhaled nebulized sodium nitrite solution to treat heart failure with preserved ejection fraction (HFpEF) currently in Phase 2 development. |
Select Development Milestones
| • | Completing negotiations with the U.S. Food and Drug Administration (FDA) on the requirements for a pivotal clinical study of Molgradex in the U.S. in Q2/2017; |
| • | Initiating a pivotal Phase 3 study of AeroVanc in Q3/2017; |
| • | Announcing an indication expansion strategy of Molgradex for the treatment of a rare lung infection in Q3/2017; |
| • | Announcing top-line results from a Phase 2/3 study of Molgradex, expected to be registration-enabling in Europe and Japan, in Q1/2018; and |
| • | Announcing results from an ongoing Phase 2 study of Aironite being conducted by the Heart Failure Clinical Research Network in H1/2018. |
Conference Call and Webcast
Savara will hold a conference call on Tuesday May 2nd, 2017, at 4:30 p.m. Eastern Time / 3:30 p.m. Central Time to provide an overview and business update. Interested parties may access the conference call by dialing (855) 239-3120 from the U.S., (855) 669-9657 from Canada, and (412) 542-4127 from outside the U.S. and should request the Savara Inc. Call. A live webcast of the conference call will be available online from the Investors section of Savara’s website at http://www.savarapharma.com/investors/events/. Replays of the webcast will be available on Savara’s website for 30 days and a telephone replay will be available through May 9th, 2017 by dialing (877) 344-7529 from the U.S., (855) 669-9658 from Canada, and (412) 317-0088 from elsewhere outside the U.S. and entering replay access code 10104600.
About Savara
Savara Inc. is a clinical-stage specialty pharmaceutical company focused on the development and commercialization of novel therapies for the treatment of serious or life-threatening rare respiratory diseases. Savara’s pipeline comprises AeroVanc, a Phase 3 ready inhaled vancomycin, Molgradex, a Phase 2/3 stage inhaled granulocyte-macrophage colony-stimulating factor, or GM-CSF and Aironite, an inhaled nebulized sodium nitrite solution to treat HFpEF. Savara’s strategy involves expanding its pipeline of best-in-class products through indication expansion, strategic development partnerships and product acquisitions, with the goal of becoming a leading company in its field. Savara’s management team has significant experience in orphan drug development and pulmonary medicine, in identifying unmet needs, creating and acquiring new product candidates, and effectively advancing them to approvals and commercialization. More information can be found at www.savarapharma.com. (Twitter: @SavaraPharma)
Forward Looking Statements
Savara cautions you that statements in this press release that are not a description of historical fact are forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. Forward-looking statements may be identified by the use of words referencing future events or circumstances such as “expect,” “intend,” “plan,” “anticipate,” “believe,” and “will,” among others. Such statements include, but are not limited to, statements regarding dynamically pursuing opportunities to develop impactful products, Savara presenting an attractive business opportunity with a pipeline of unique products with considerable market potential, as well as significant value-driving clinical milestones, Savara’s intent to continue its growth strategy focused on indication expansion, strategic development partnerships and product acquisitions, Savara’s pipeline and select developmental milestones. Savara may not actually achieve any of the matters referred to in such forward looking
statements, and you should not place undue reliance on these forward-looking statements. Because such statements are subject to risks and uncertainties, actual results may differ materially from those expressed or implied by such forward-looking statements. These forward-looking statements are based upon Savara’s current expectations and involve assumptions that may never materialize or may prove to be incorrect. Actual results and the timing of events could differ materially from those anticipated in such forward-looking statements as a result of various risks and uncertainties, which include, without limitation, risks and uncertainties associated with the ability to project future cash utilization and reserves needed for contingent future liabilities and business operations, the availability of sufficient resources for Savara’s operations and to conduct or continue planned clinical development programs, the timing and ability of Savara to raise additional equity capital to fund continued operations; the ability to successfully develop Savara’s product candidates, and the risks associated with the process of developing, obtaining regulatory approval for and commercializing drug candidates that are safe and effective for use as human therapeutics. Risks and uncertainties facing Savara are described more fully in Savara’s filings with the Securities and Exchange Commission including the most recent Form 8-K filed on April 27, 2017, other filings on Form 8-K, the Annual Report on Form 10-K for the fiscal year ended December 31, 2016 and the Registration Statement on Form S-4 related to the Mast/Savara merger. You are cautioned not to place undue reliance on forward-looking statements, which speak only as of the date on which they were made. Savara undertakes any obligation to update such statements to reflect events that occur or circumstances that exist after the date on which they were made, except as may be required by law.
Contact:
Savara Inc.
Ioana C. Hone (ir@savarapharma.com)
(512) 961-1891
###

Email Alerts
Enter your e-mail address to receive alerts whenever certain new company information is posted to this site.